

Novel Insights into Alcoholic Liver Disease: Iron Overload, Iron Sensing and Hemolysis
- PMID: 35959455
- PMCID: PMC9328032
- DOI: 10.2478/jtim-2021-0056
Novel Insights into Alcoholic Liver Disease: Iron Overload, Iron Sensing and Hemolysis
Abstract
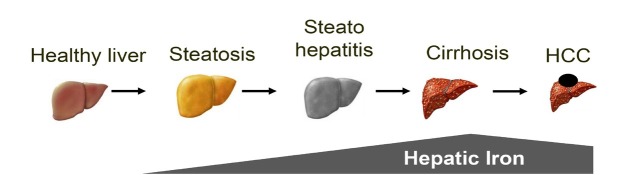
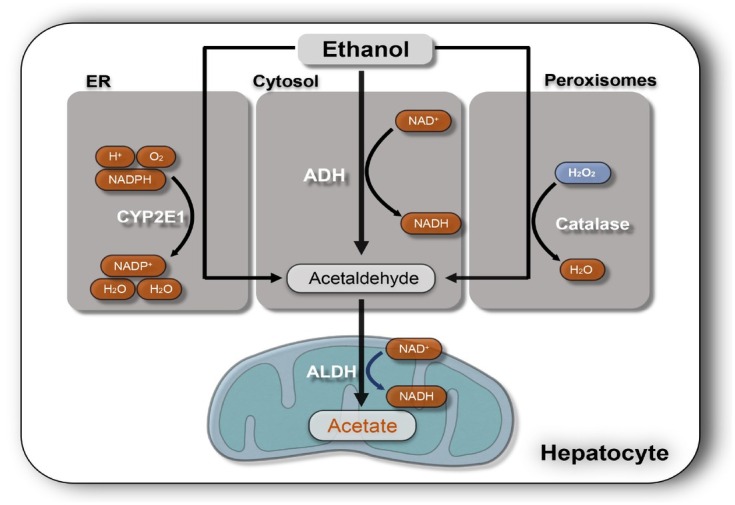
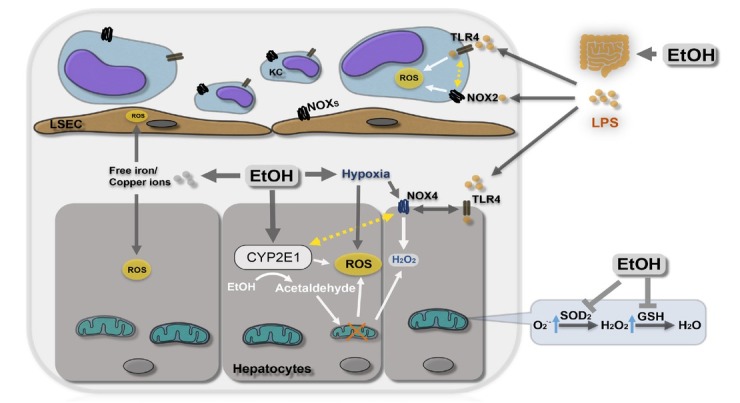
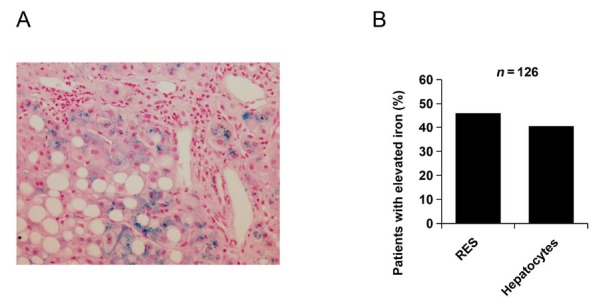
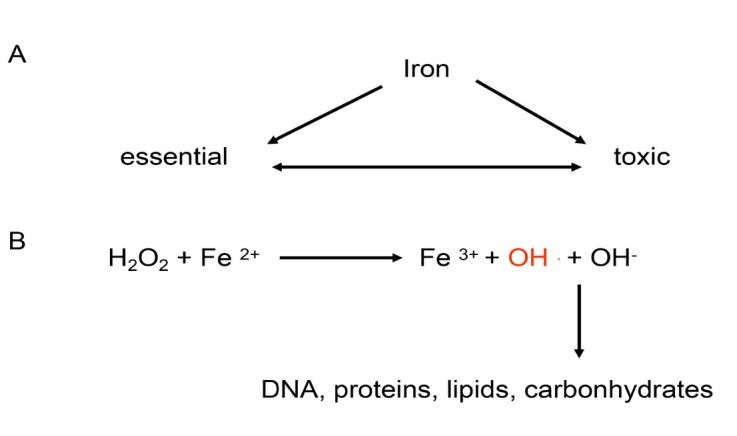
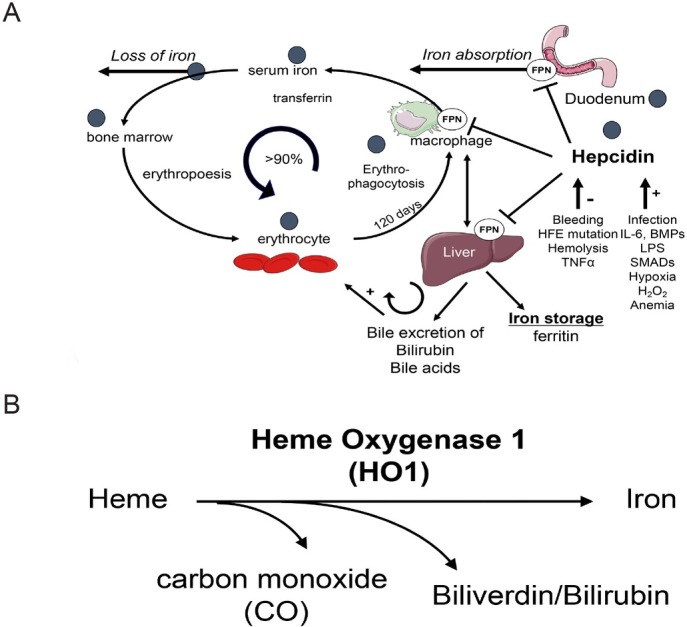
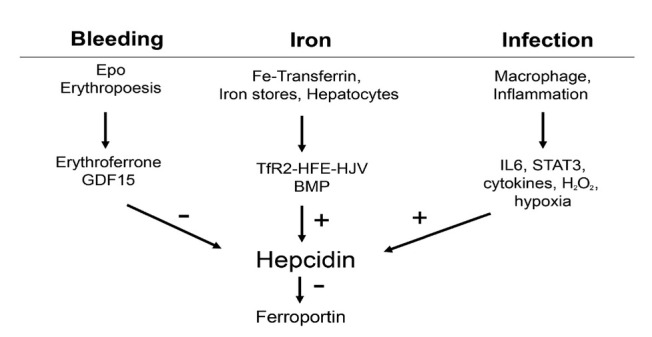
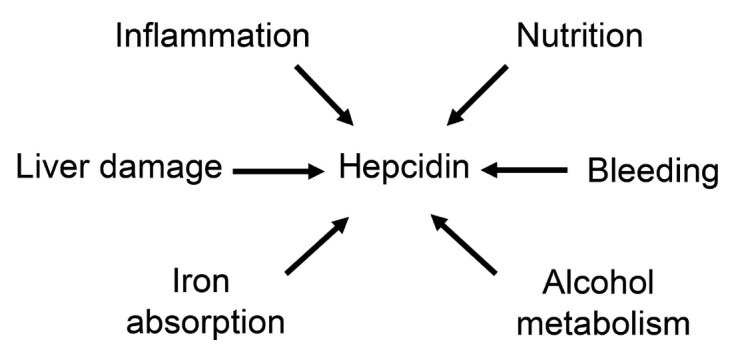
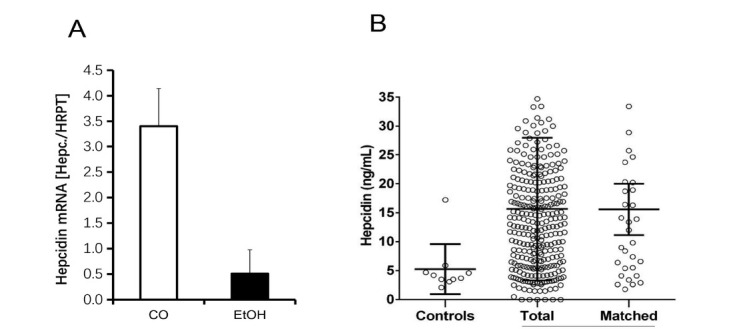
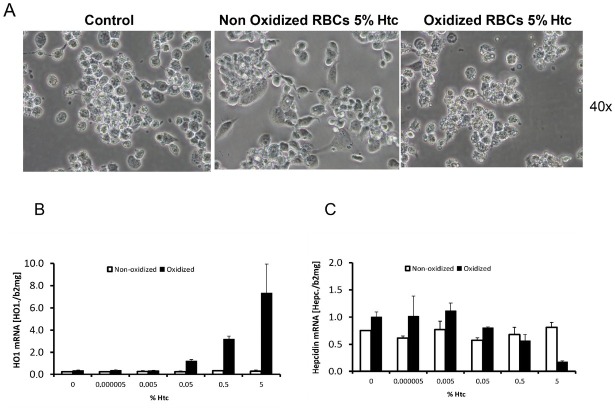
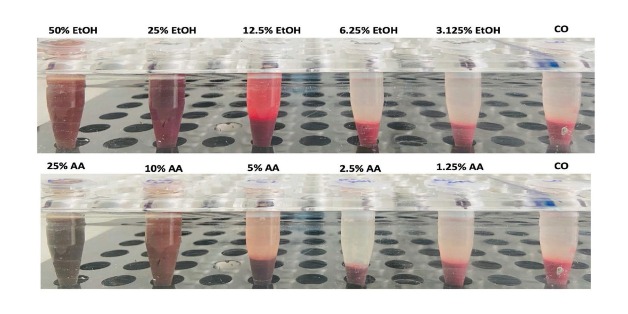
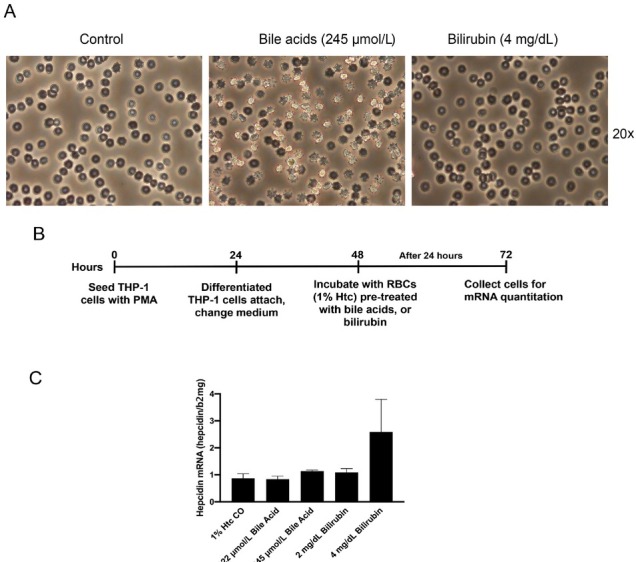
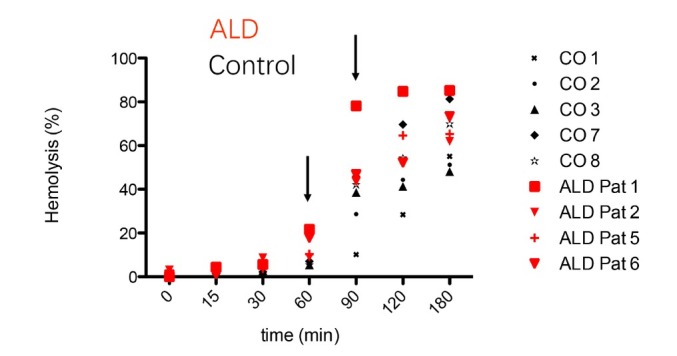
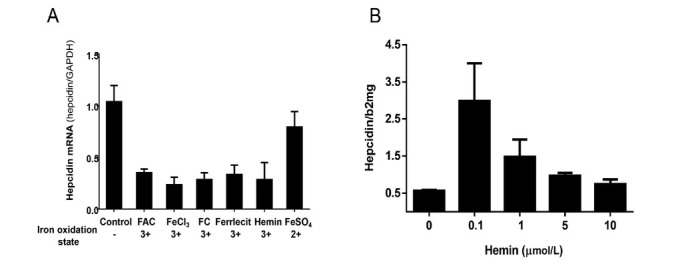
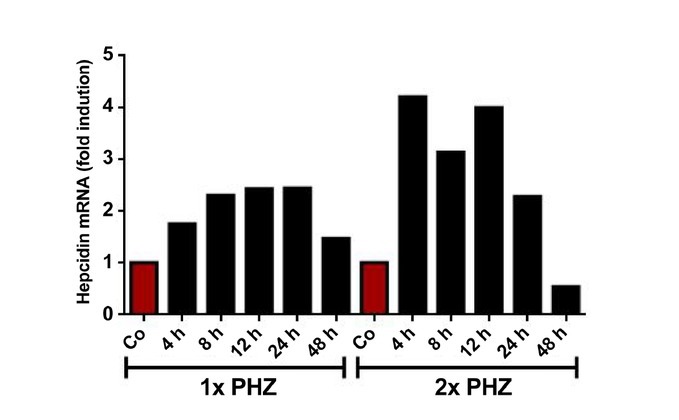
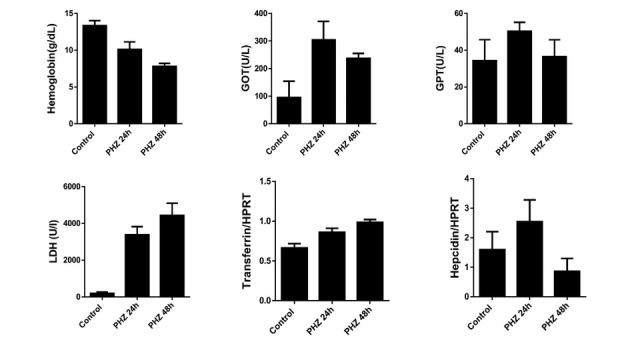
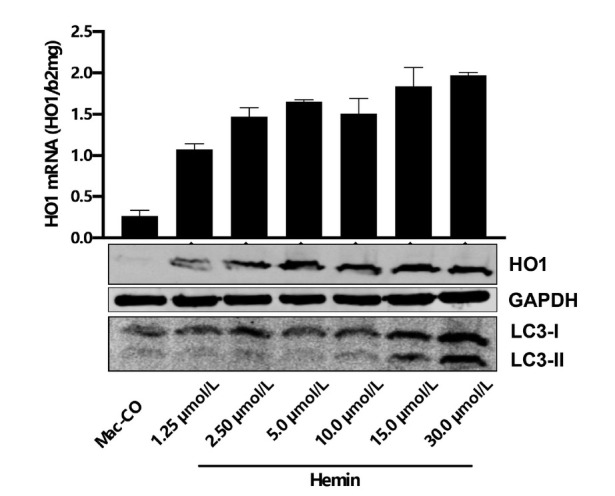
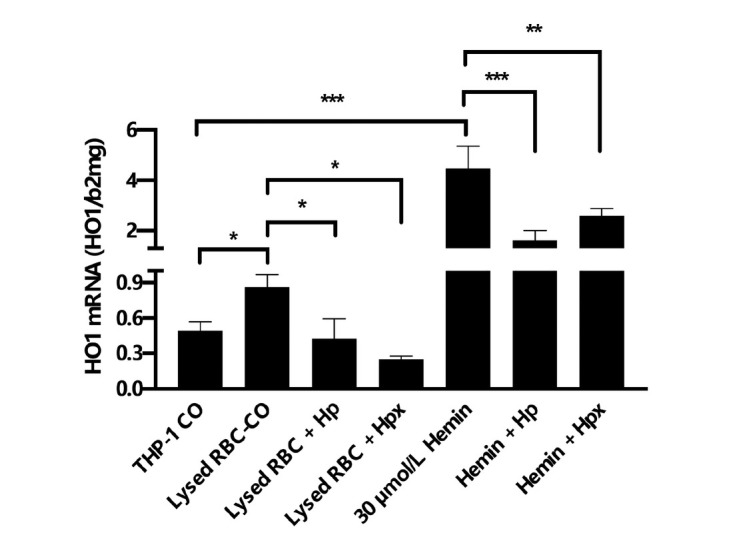
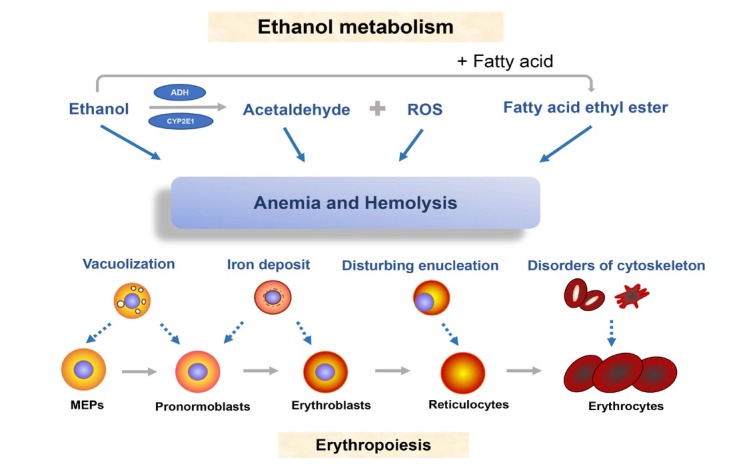
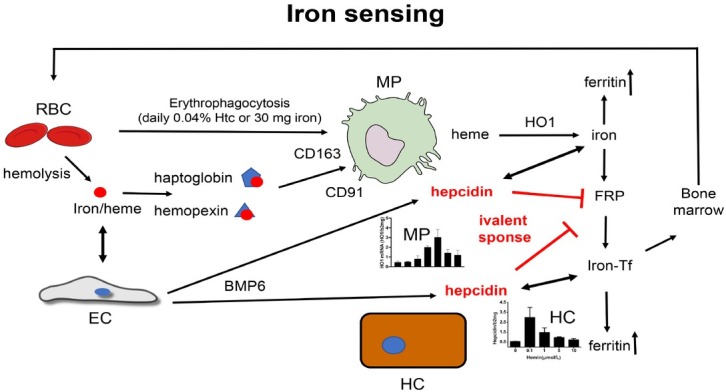
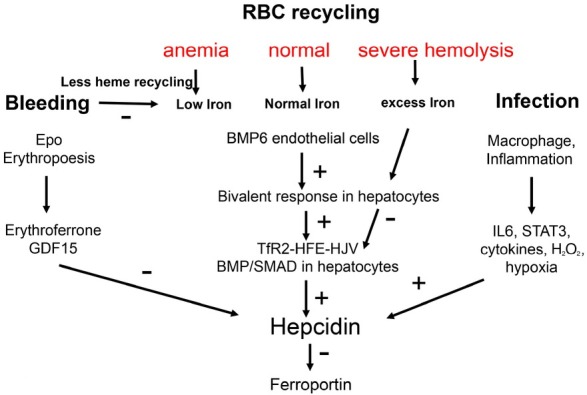
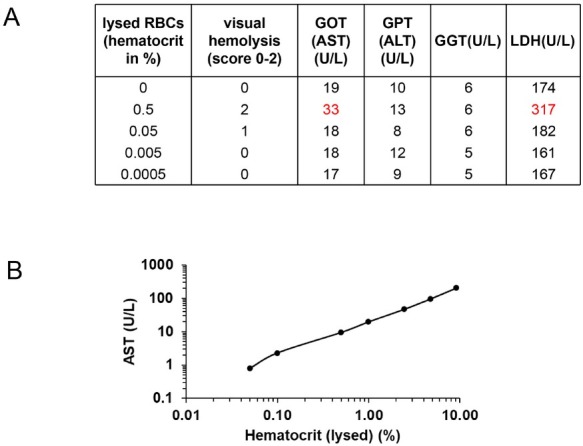
The liver is the major target organ of continued alcohol consumption at risk and resulting alcoholic liver disease (ALD) is the most common liver disease worldwide. The underlying molecular mechanisms are still poorly understood despite decades of scientific effort limiting our abilities to identify those individuals who are at risk to develop the disease, to develop appropriate screening strategies and, in addition, to develop targeted therapeutic approaches. ALD is predestined for the newly evolving translational medicine, as conventional clinical and health care structures seem to be constrained to fully appreciate this disease. This concept paper aims at summarizing the 15 years translational experience at the Center of Alcohol Research in Heidelberg, namely based on the long-term prospective and detailed characterization of heavy drinkers with mortality data. In addition, novel experimental findings will be presented. A special focus will be the long-known hepatic iron accumulation, the somewhat overlooked role of the hematopoietic system and novel insights into iron sensing and the role of hepcidin. Our preliminary work indicates that enhanced red blood cell (RBC) turnover is critical for survival in ALD patients. RBC turnover is not primarily due to vitamin deficiency but rather to ethanol toxicity directly targeted to erythrocytes but also to the bone marrow stem cell compartment. These novel insights also help to explain long-known aspects of ALD such as mean corpuscular volume of erythrocytes (MCV) and elevated aspartate transaminase (GOT/AST) levels. This work also aims at identifying future projects, naming unresolved observations, and presenting novel hypothetical concepts still requiring future validation.
Keywords: alcoholic liver disease; cd163; erythrophagocytosis; hemolysis; hepcidin; iron overload; red blood cell.
© 2022 Sebastian Mueller, Cheng Chen, Johannes Mueller, Shijin Wang, published by Sciendo.
Conflict of interest statement
Conflict of Interest None declared.
Figures
References
-
- Ofliver E. EASL Clinical Practice Guidelines: Management of alcohol-related liver disease. J Hepatol. 2018;69:154–81. - PubMed
-
- Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology. 2015;148:547–55. et al. - PubMed
-
- Burra P, Senzolo M, Adam R, Delvart V, Karam V, Germani G. Liver transplantation for alcoholic liver disease in Europe: a study from the ELTR (European Liver Transplant Registry) Am J Transplant. 2010;10:138–48. et al. - PubMed
-
- Teli MR, Day CP, Burt AD, Bennett MK, James OF. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet. 1995;346:987–90. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials