

Amyloid precursor protein (APP) and amyloid β (Aβ) interact with cell adhesion molecules: Implications in Alzheimer's disease and normal physiology
- PMID: 35959488
- PMCID: PMC9360506
- DOI: 10.3389/fcell.2022.969547
Amyloid precursor protein (APP) and amyloid β (Aβ) interact with cell adhesion molecules: Implications in Alzheimer's disease and normal physiology
Abstract
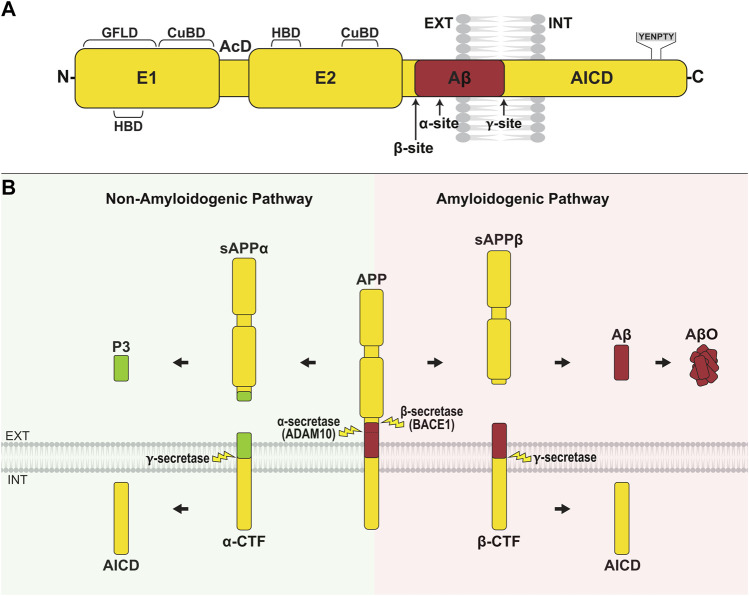
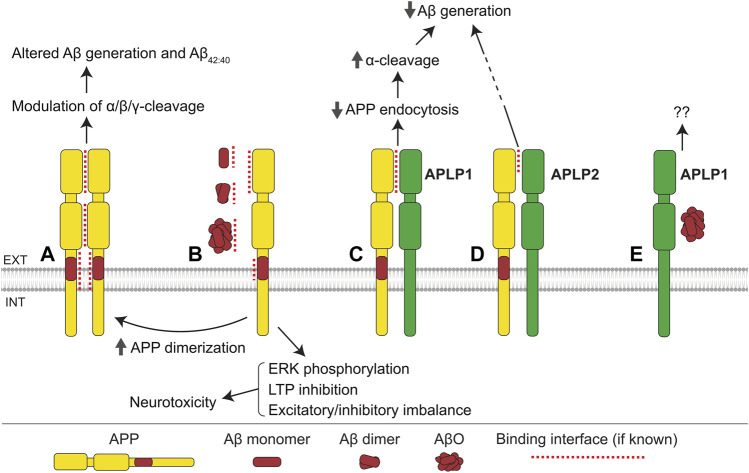
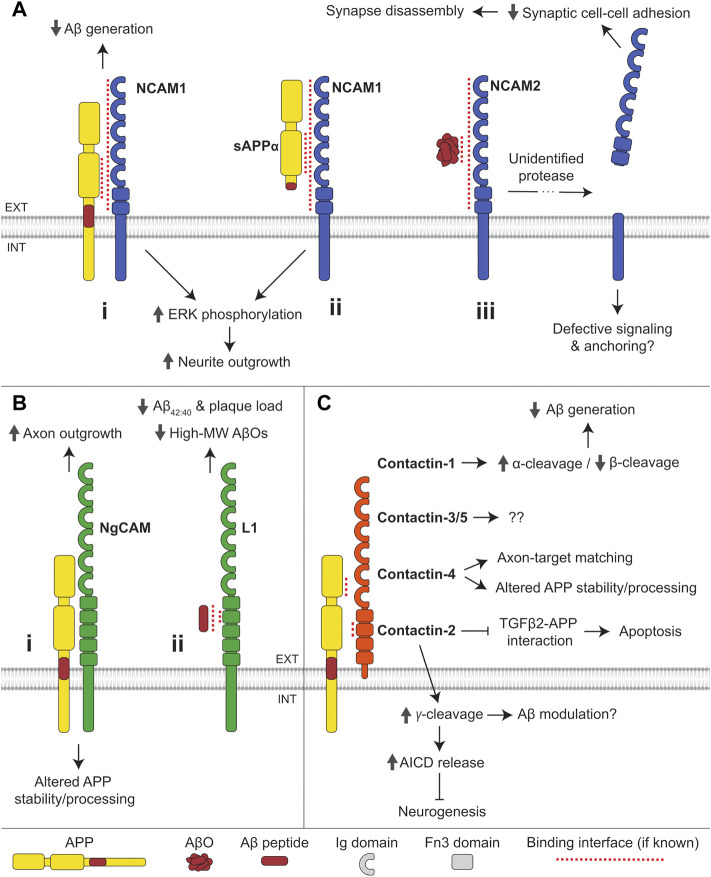
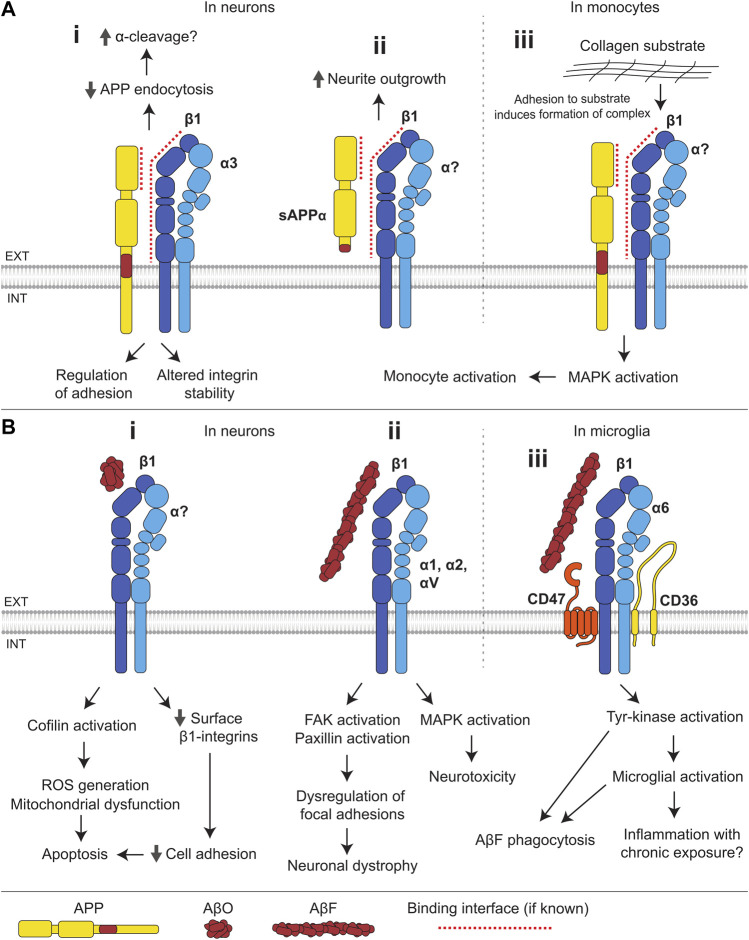
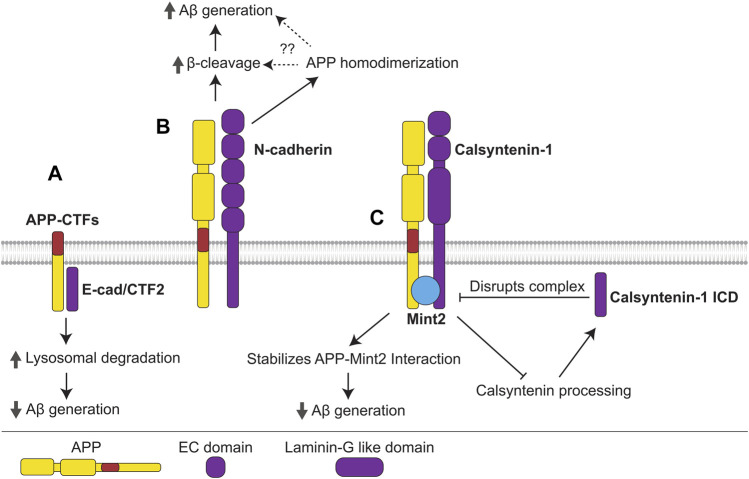
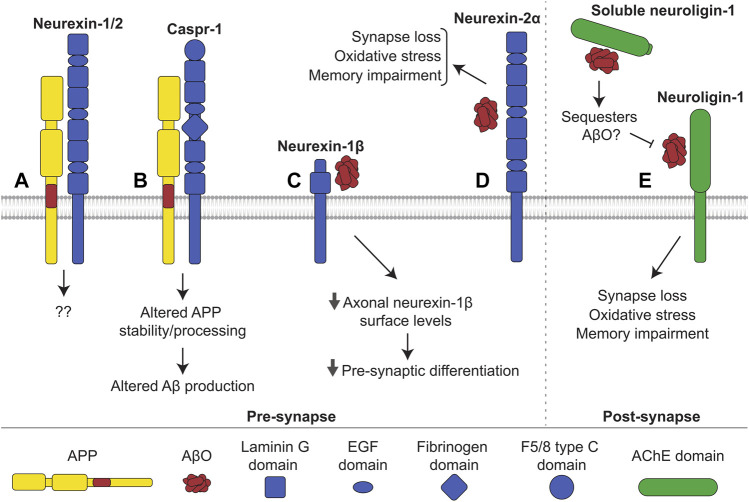
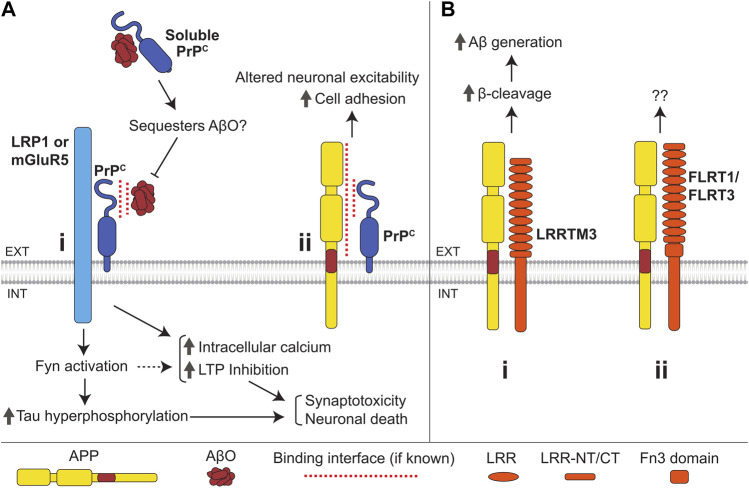
Alzheimer's disease (AD) is an incurable neurodegenerative disorder in which dysfunction and loss of synapses and neurons lead to cognitive impairment and death. Accumulation and aggregation of neurotoxic amyloid-β (Aβ) peptides generated via amyloidogenic processing of amyloid precursor protein (APP) is considered to play a central role in the disease etiology. APP interacts with cell adhesion molecules, which influence the normal physiological functions of APP, its amyloidogenic and non-amyloidogenic processing, and formation of Aβ aggregates. These cell surface glycoproteins also mediate attachment of Aβ to the neuronal cell surface and induce intracellular signaling contributing to Aβ toxicity. In this review, we discuss the current knowledge surrounding the interactions of cell adhesion molecules with APP and Aβ and analyze the evidence of the critical role these proteins play in regulating the processing and physiological function of APP as well as Aβ toxicity. This is a necessary piece of the complex AD puzzle, which we should understand in order to develop safe and effective therapeutic interventions for AD.
Keywords: Alzheimer’s disease; amyloid precursor protein (APP); amyloid-beta; cell adhesion molecule (CAM); immunoglobulin superfamily; integrin; neurexin; prion protein (PrP).
Copyright © 2022 Pfundstein, Nikonenko and Sytnyk.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Alzheimer’s Association (2022). 2022 Alzheimer's disease facts and figures. Alzheimer's Dementia 18, 700–789. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials