

Loss of mitochondrial fatty acid β-oxidation protein short-chain Enoyl-CoA hydratase disrupts oxidative phosphorylation protein complex stability and function
- PMID: 35962613
- PMCID: PMC10087869
- DOI: 10.1111/febs.16595
Loss of mitochondrial fatty acid β-oxidation protein short-chain Enoyl-CoA hydratase disrupts oxidative phosphorylation protein complex stability and function
Abstract
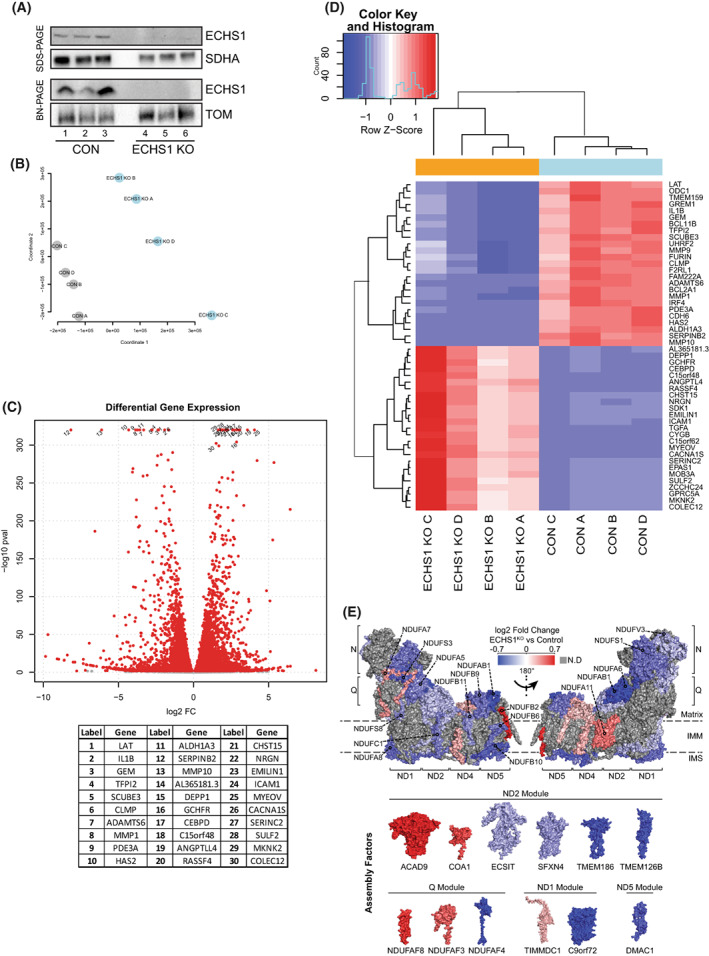
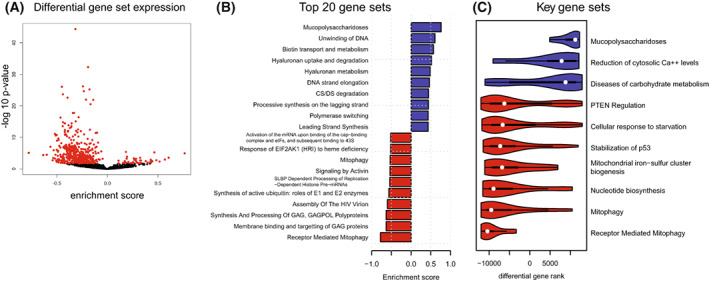
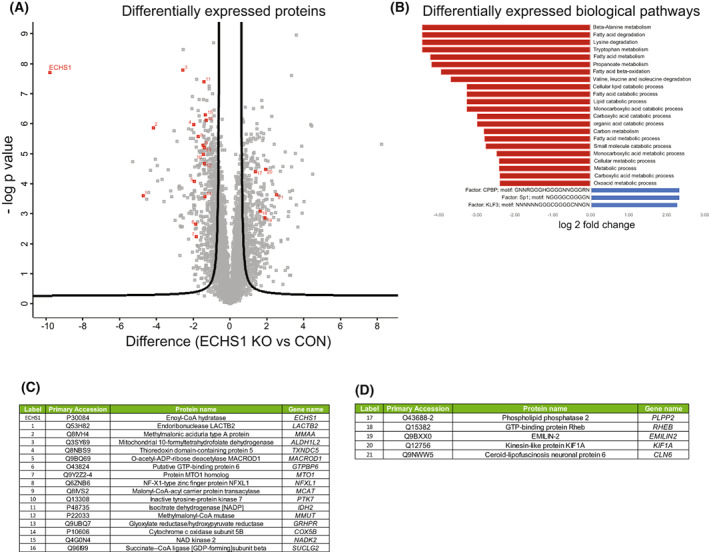
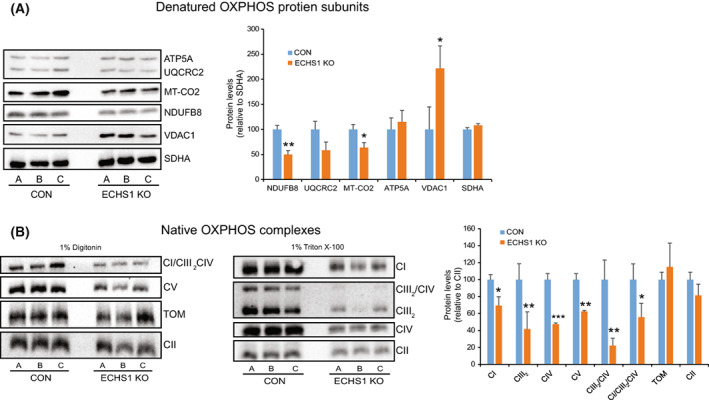
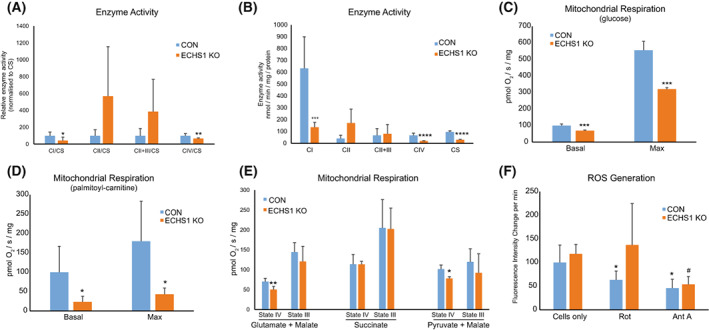
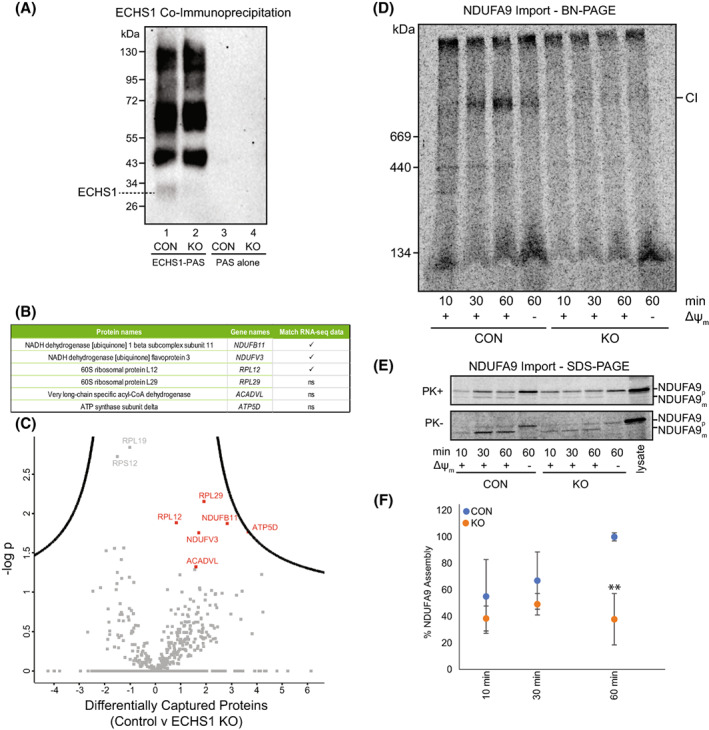
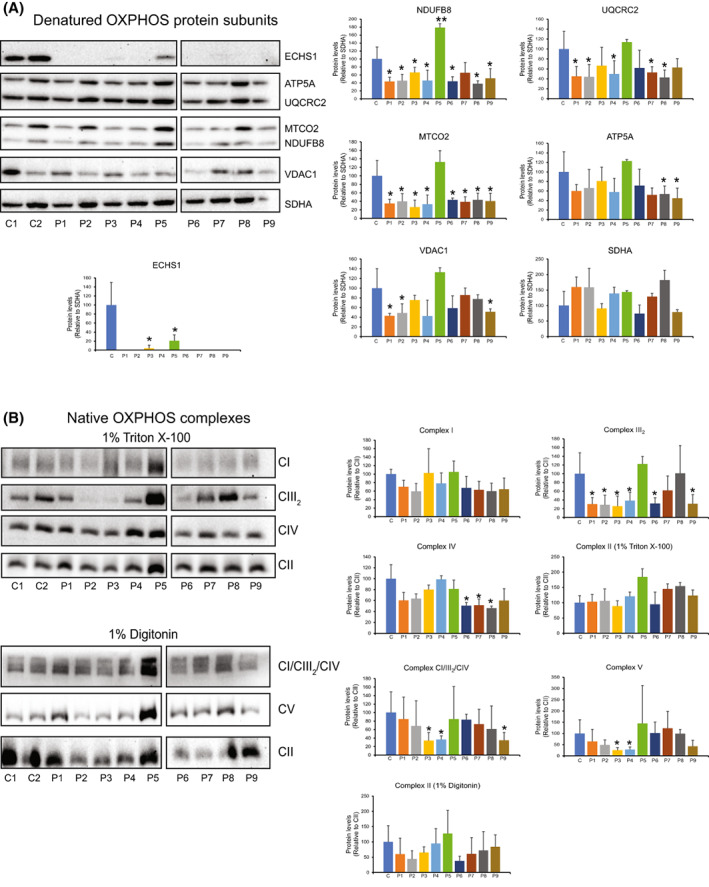
Short-chain enoyl-CoA hydratase 1 (ECHS1) is involved in the second step of mitochondrial fatty acid β-oxidation (FAO), catalysing the hydration of short-chain enoyl-CoA esters to short-chain 3-hyroxyl-CoA esters. Genetic deficiency in ECHS1 (ECHS1D) is associated with a specific subset of Leigh Syndrome, a disease typically caused by defects in oxidative phosphorylation (OXPHOS). Here, we examined the molecular pathogenesis of ECHS1D using a CRISPR/Cas9 edited human cell 'knockout' model and fibroblasts from ECHS1D patients. Transcriptome analysis of ECHS1 'knockout' cells showed reductions in key mitochondrial pathways, including the tricarboxylic acid cycle, receptor-mediated mitophagy and nucleotide biosynthesis. Subsequent proteomic analyses confirmed these reductions and revealed additional defects in mitochondrial oxidoreductase activity and fatty acid β-oxidation. Functional analysis of ECHS1 'knockout' cells showed reduced mitochondrial oxygen consumption rates when metabolising glucose or OXPHOS complex I-linked substrates, as well as decreased complex I and complex IV enzyme activities. ECHS1 'knockout' cells also exhibited decreased OXPHOS protein complex steady-state levels (complex I, complex III2 , complex IV, complex V and supercomplexes CIII2 /CIV and CI/CIII2 /CIV), which were associated with a defect in complex I assembly. Patient fibroblasts exhibit varied reduction of mature OXPHOS complex steady-state levels, with defects detected in CIII2 , CIV, CV and the CI/CIII2 /CIV supercomplex. Overall, these findings highlight the contribution of defective OXPHOS function, in particular complex I deficiency, to the molecular pathogenesis of ECHS1D.
Keywords: ECHS1 deficiency; OXPHOS; fatty acid oxidation; mitochondria; mitochondrial disease; short-chain enoyl-CoA hydratase.
© 2022 The Authors. The FEBS Journal published by John Wiley & Sons Ltd on behalf of Federation of European Biochemical Societies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Stimulating Mitochondrial Biogenesis with Deoxyribonucleosides Increases Functional Capacity in ECHS1-Deficient Cells.Int J Mol Sci. 2022 Oct 20;23(20):12610. doi: 10.3390/ijms232012610. Int J Mol Sci. 2022. PMID: 36293464 Free PMC article.
-
Understanding the role of OXPHOS dysfunction in the pathogenesis of ECHS1 deficiency.FEBS Lett. 2020 Feb;594(4):590-610. doi: 10.1002/1873-3468.13735. Epub 2020 Jan 29. FEBS Lett. 2020. PMID: 31944285 Review.
-
Mitochondrial Fatty Acid Oxidation Disorders Associated with Short-Chain Enoyl-CoA Hydratase (ECHS1) Deficiency.Cells. 2018 May 23;7(6):46. doi: 10.3390/cells7060046. Cells. 2018. PMID: 29882869 Free PMC article. Review.
-
Structural and biochemical mechanism of short-chain enoyl-CoA hydratase (ECHS1) substrate recognition.Commun Biol. 2025 Apr 16;8(1):619. doi: 10.1038/s42003-025-07924-0. Commun Biol. 2025. PMID: 40240482 Free PMC article.
-
Molecular and in silico investigation of a novel ECHS1 gene mutation in a consanguine family with short-chain enoyl-CoA hydratase deficiency and Mt-DNA depletion: effect on trimer assembly and catalytic activity.Metab Brain Dis. 2024 Apr;39(4):611-623. doi: 10.1007/s11011-024-01343-6. Epub 2024 Feb 16. Metab Brain Dis. 2024. PMID: 38363494
Cited by
-
Time-resolved mitochondrial screen identifies regulatory components of oxidative metabolism.EMBO Rep. 2025 Jun;26(12):3045-3074. doi: 10.1038/s44319-025-00459-9. Epub 2025 Apr 29. EMBO Rep. 2025. PMID: 40301572 Free PMC article.
-
Acetylation of proximal cysteine-lysine pairs by alcohol metabolism.Redox Biol. 2025 Feb;79:103462. doi: 10.1016/j.redox.2024.103462. Epub 2024 Dec 12. Redox Biol. 2025. PMID: 39729908 Free PMC article.
-
Valine and Inflammation Drive Epilepsy in a Mouse Model of ECHS1 Deficiency.bioRxiv [Preprint]. 2024 Jun 13:2024.06.13.598697. doi: 10.1101/2024.06.13.598697. bioRxiv. 2024. PMID: 38915588 Free PMC article. Preprint.
-
The Pathogenesis of Very Long-Chain Acyl-CoA Dehydrogenase Deficiency.Biomolecules. 2025 Mar 14;15(3):416. doi: 10.3390/biom15030416. Biomolecules. 2025. PMID: 40149952 Free PMC article. Review.
-
Echs1-mediated histone crotonylation facilitates zygotic genome activation and expression of repetitive elements in early mammalian embryos.Nat Commun. 2025 Jul 1;16(1):5630. doi: 10.1038/s41467-025-60565-z. Nat Commun. 2025. PMID: 40593492 Free PMC article.
References
-
- Burgin HJ, McKenzie M. Understanding the role of OXPHOS dysfunction in the pathogenesis of ECHS1 deficiency. FEBS Lett. 2020;594:590–610. - PubMed
-
- Peters H, Buck N, Wanders R, Ruiter J, Waterham H, Koster J, et al. ECHS1 mutations in Leigh disease: a new inborn error of metabolism affecting valine metabolism. Brain. 2014;137:2903–8. - PubMed
-
- Lake NJ, Compton AG, Rahman S, Thorburn DR. Leigh syndrome: one disorder, more than 75 monogenic causes. Ann Neurol. 2016;79:190–203. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
