

Evaluation of various distance computation methods for construction of haplotype-based phylogenies from large MLST datasets
- PMID: 35963590
- PMCID: PMC10127246
- DOI: 10.1016/j.ympev.2022.107608
Evaluation of various distance computation methods for construction of haplotype-based phylogenies from large MLST datasets
Abstract
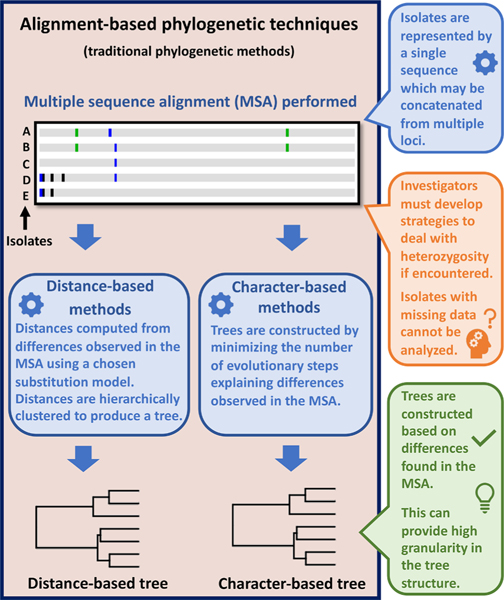
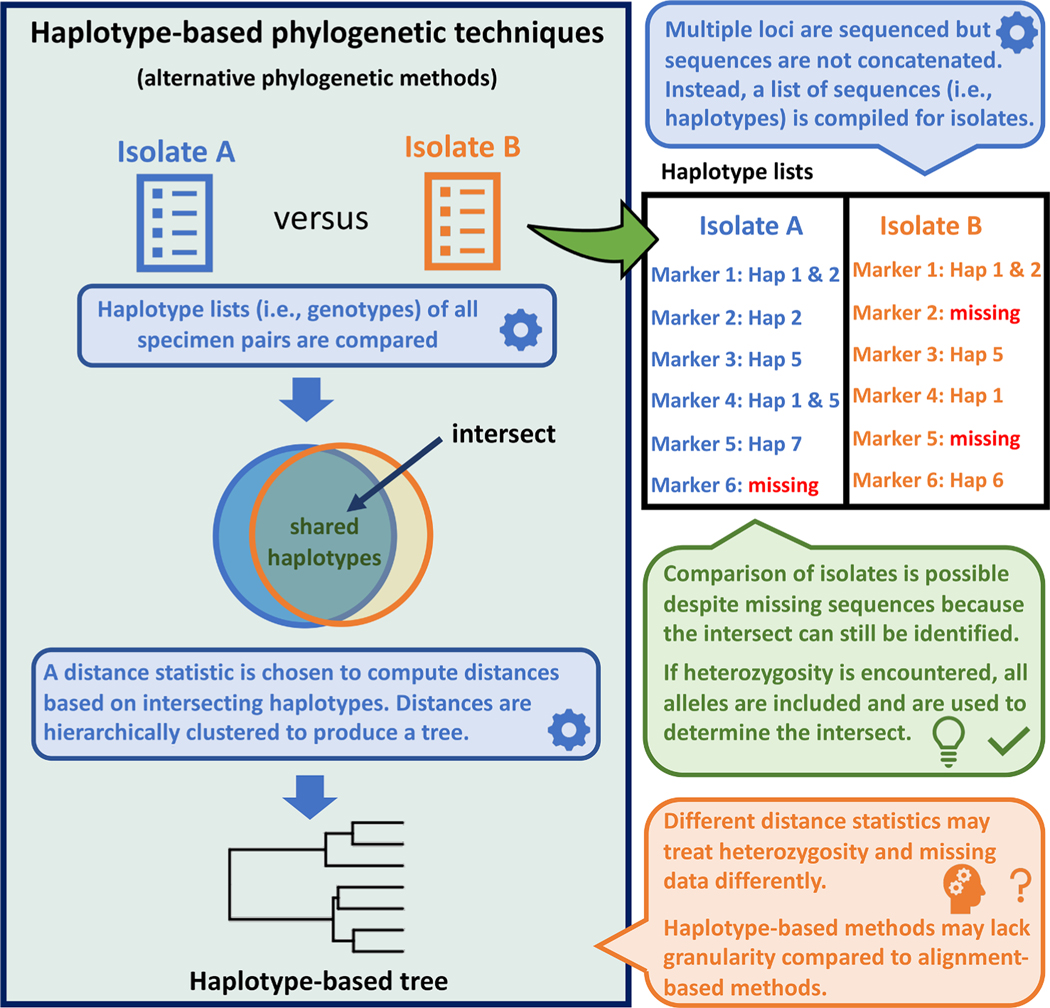
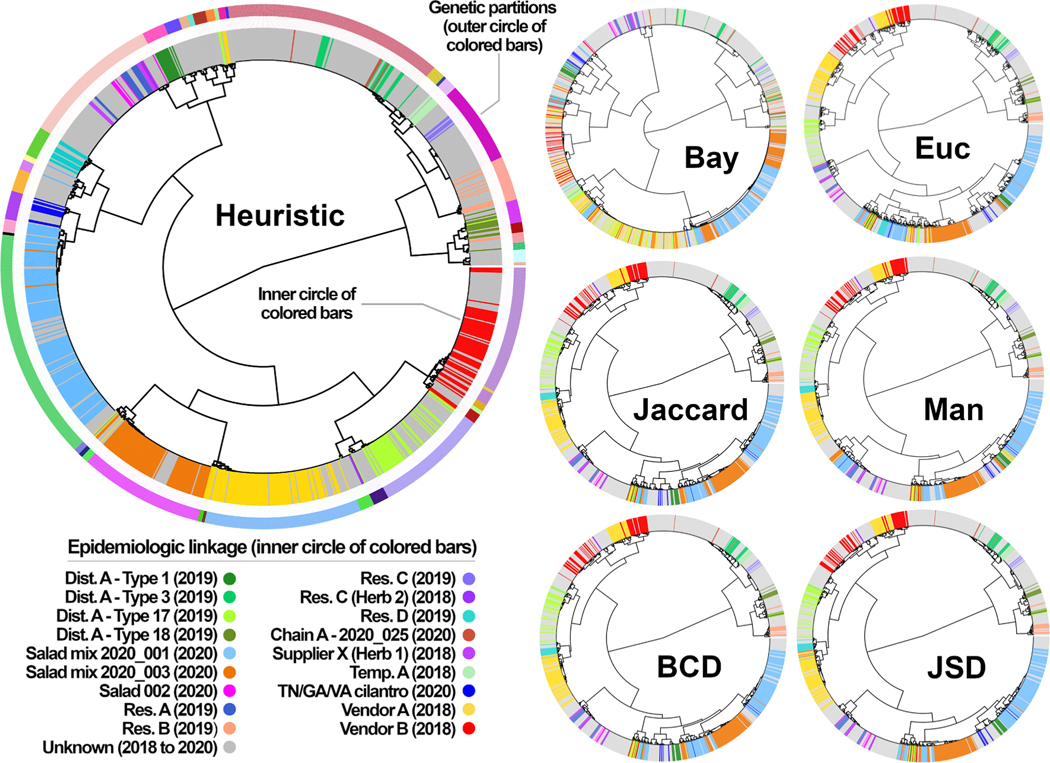
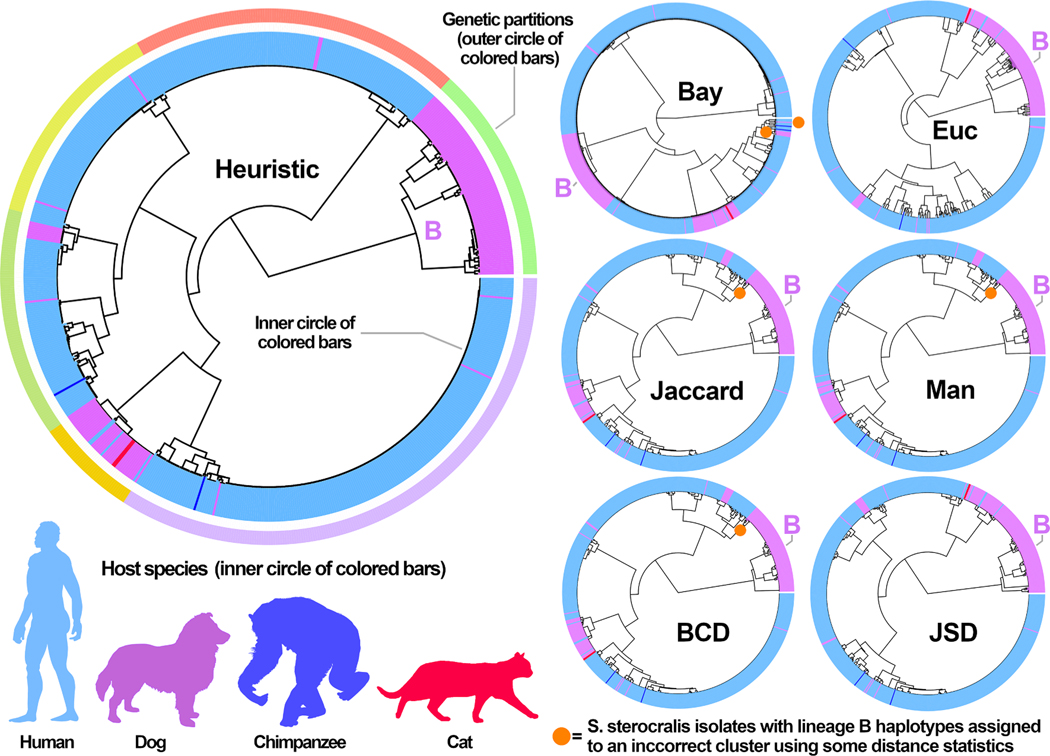
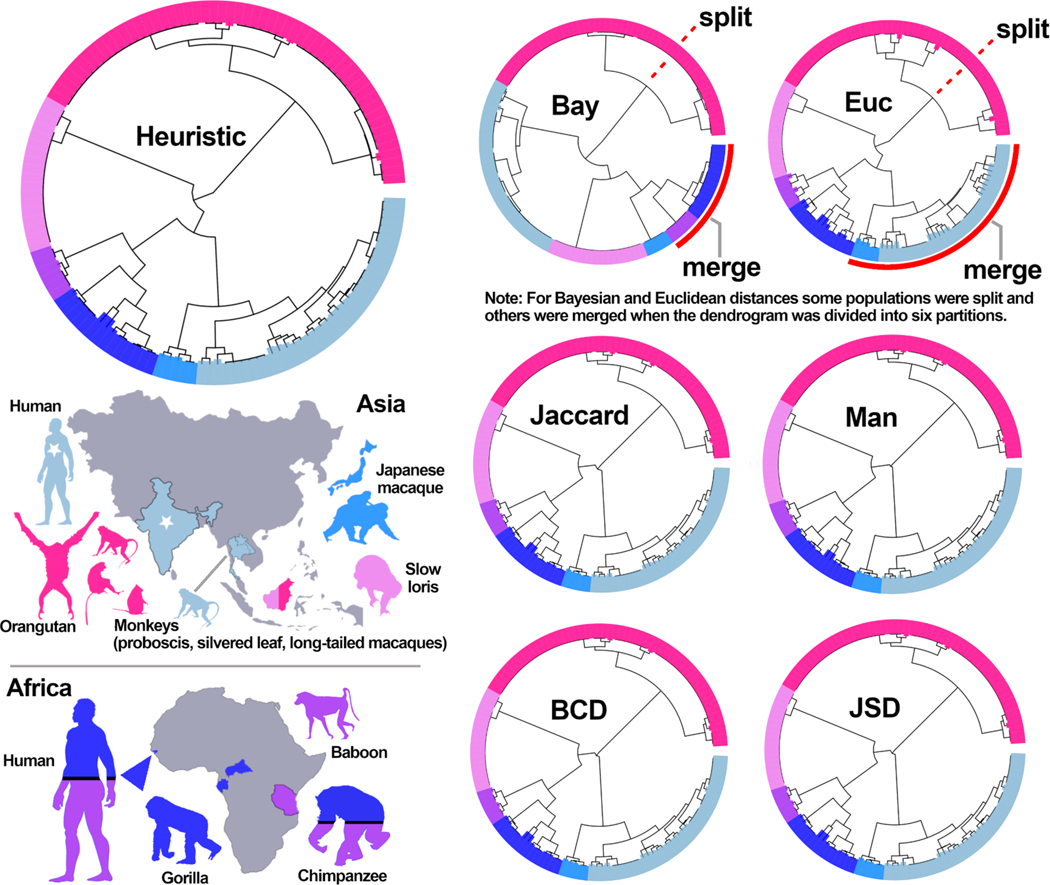
Multi-locus sequence typing (MLST) is widely used to investigate genetic relationships among eukaryotic taxa, including parasitic pathogens. MLST analysis workflows typically involve construction of alignment-based phylogenetic trees - i.e., where tree structures are computed from nucleotide differences observed in a multiple sequence alignment (MSA). Notably, alignment-based phylogenetic methods require that all isolates/taxa are represented by a single sequence. When multiple loci are sequenced these sequences may be concatenated to produce one tree that includes information from all loci. Alignment-based phylogenetic techniques are robust and widely used yet possess some shortcomings, including how heterozygous sites are handled, intolerance for missing data (i.e., partial genotypes), and differences in the way insertions-deletions (indels) are scored/treated during tree construction. In certain contexts, 'haplotype-based' methods may represent a viable alternative to alignment-based techniques, as they do not possess the aforementioned limitations. This is namely because haplotype-based methods assess genetic similarity based on numbers of shared (i.e., intersecting) haplotypes as opposed to similarities in nucleotide composition observed in an MSA. For haplotype-based comparisons, choosing an appropriate distance statistic is fundamental, and several statistics are available to choose from. However, a comprehensive assessment of various available statistics for their ability to produce a robust haplotype-based phylogenetic reconstruction has not yet been performed. We evaluated seven distance statistics by applying them to extant MLST datasets from the gastrointestinal parasite Cyclospora cayetanensis and two species of pathogenic nematode of the genus Strongyloides. We compare the genetic relationships identified using each statistic to epidemiologic, geographic, and host metadata. We show that Barratt's heuristic definition of genetic distance was the most robust among the statistics evaluated. Consequently, it is proposed that Barratt's heuristic represents a useful approach for use in the context of challenging MLST datasets possessing features (i.e., high heterozygosity, partial genotypes, and indel or repeat-based polymorphisms) that confound or preclude the use of alignment-based methods.
Keywords: Cyclospora; Eukaryotes; Genotyping; Parasites; Phylogeny; Strongyloides.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of Competing Interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
-
- Anonymous. 2018. Domestically Acquired Cases of Cyclosporiasis — United States, May–August 2018. Centers for Disease Control and Prevention; 2018 [cited 2020]; Available from: https://www.cdc.gov/parasites/cyclosporiasis/outbreaks/2018/c-082318/ind....
-
- Anonymous. 2019a. Domestically Acquired Cases of Cyclosporiasis — United States, May–August 2019. Centers for Disease Control and Prevention; 2019 [cited 2020]; Available from: https://www.cdc.gov/parasites/cyclosporiasis/outbreaks/2019/a-050119/ind....
-
- Anonymous. 2019b. Outbreak of Cyclospora Infections Linked to Fresh Basil from Siga Logistics de RL de CV of Morelos, Mexico. Centers for Disease Control and Prevention; 2019 [cited 2020]; Available from: https://www.cdc.gov/parasites/cyclosporiasis/outbreaks/2019/weekly/index....
-
- Anonymous. 2020. Domestically Acquired Cases of Cyclosporiasis — United States, May–August 2020. Centers for Disease Control and Prevention; 2020 [cited 2021]; Available from: https://www.cdc.gov/parasites/cyclosporiasis/outbreaks/2020/seasonal/ind....
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
