

Targeted therapeutics and novel signaling pathways in non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH)
- PMID: 35963848
- PMCID: PMC9376100
- DOI: 10.1038/s41392-022-01119-3
Targeted therapeutics and novel signaling pathways in non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH)
Abstract
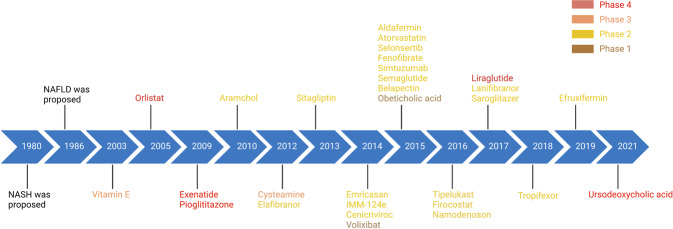
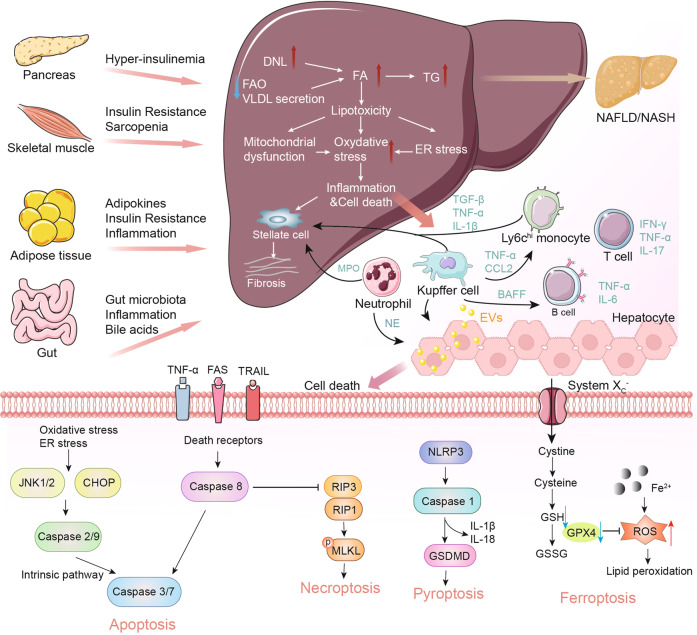
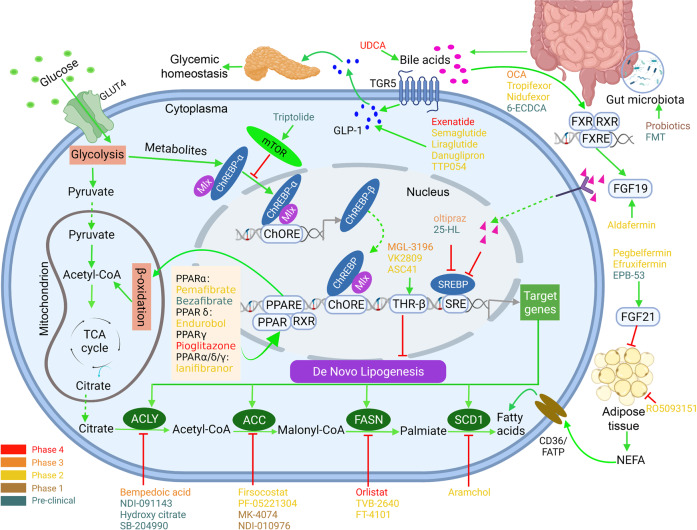
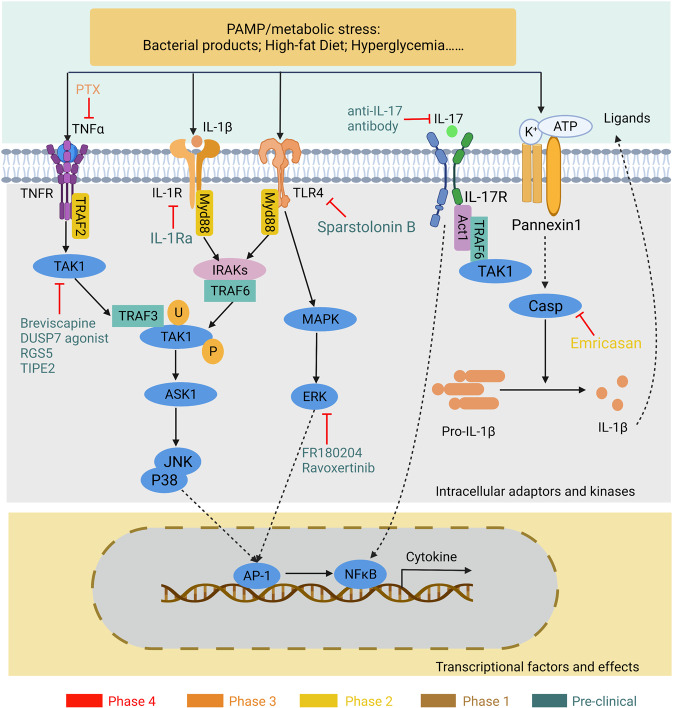
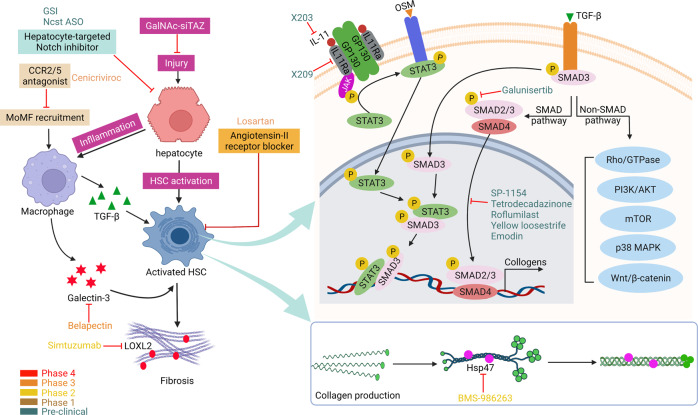
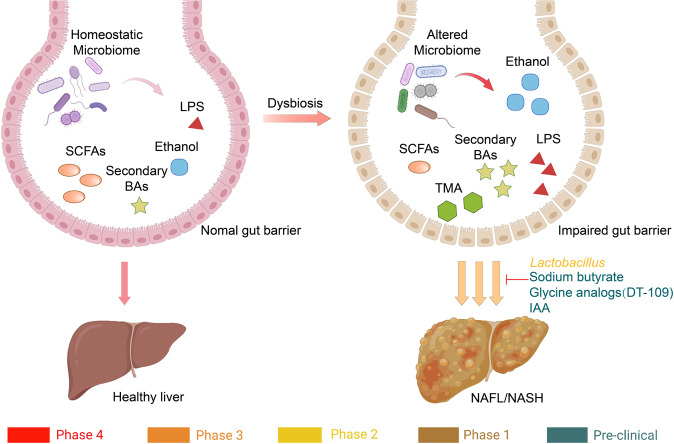
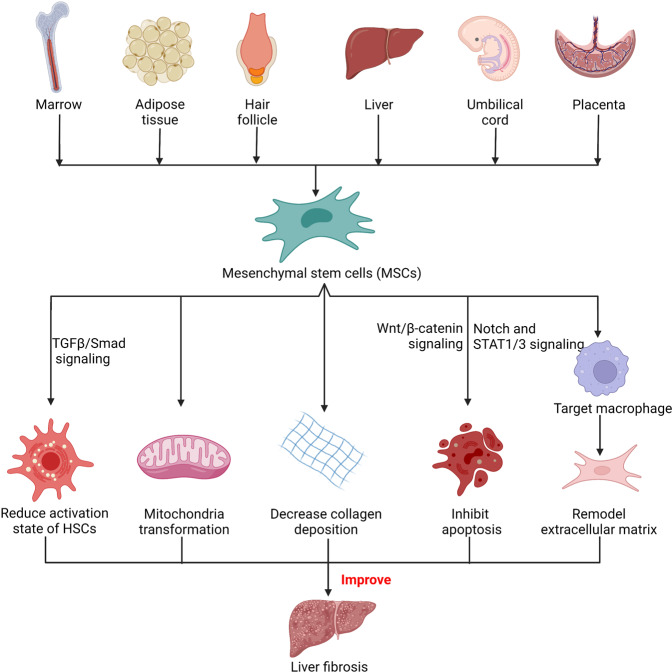
Non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH) has become the leading cause of liver disease worldwide. NASH, an advanced form of NAFL, can be progressive and more susceptible to developing cirrhosis and hepatocellular carcinoma. Currently, lifestyle interventions are the most essential and effective strategies for preventing and controlling NAFL without the development of fibrosis. While there are still limited appropriate drugs specifically to treat NAFL/NASH, growing progress is being seen in elucidating the pathogenesis and identifying therapeutic targets. In this review, we discussed recent developments in etiology and prospective therapeutic targets, as well as pharmacological candidates in pre/clinical trials and patents, with a focus on diabetes, hepatic lipid metabolism, inflammation, and fibrosis. Importantly, growing evidence elucidates that the disruption of the gut-liver axis and microbe-derived metabolites drive the pathogenesis of NAFL/NASH. Extracellular vesicles (EVs) act as a signaling mediator, resulting in lipid accumulation, macrophage and hepatic stellate cell activation, further promoting inflammation and liver fibrosis progression during the development of NAFL/NASH. Targeting gut microbiota or EVs may serve as new strategies for the treatment of NAFL/NASH. Finally, other mechanisms, such as cell therapy and genetic approaches, also have enormous therapeutic potential. Incorporating drugs with different mechanisms and personalized medicine may improve the efficacy to better benefit patients with NAFL/NASH.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
