

Evaluation of immune evasion in SARS-CoV-2 Delta and Omicron variants
- PMID: 35965661
- PMCID: PMC9359593
- DOI: 10.1016/j.csbj.2022.08.010
Evaluation of immune evasion in SARS-CoV-2 Delta and Omicron variants
Abstract
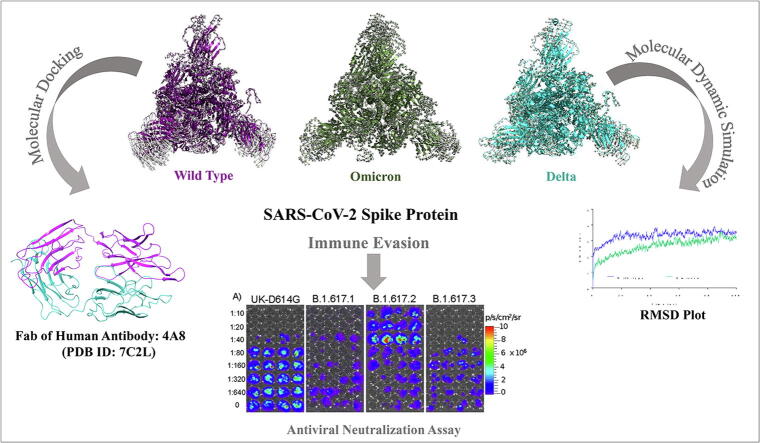
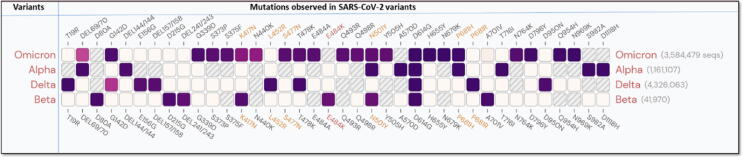
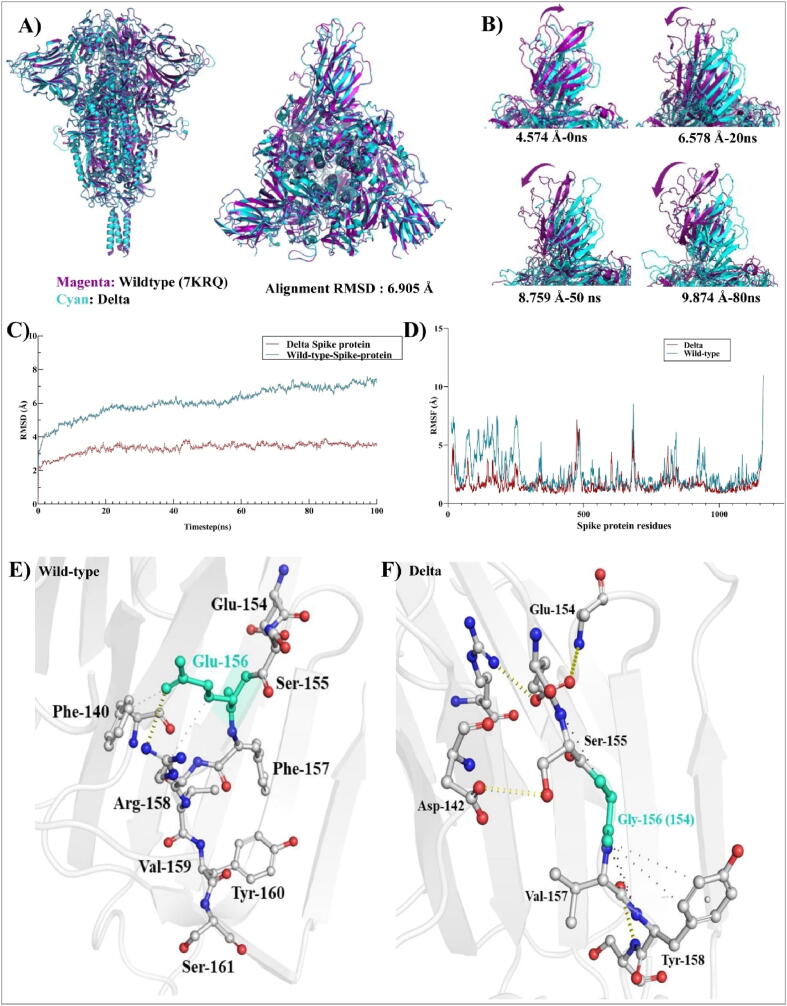
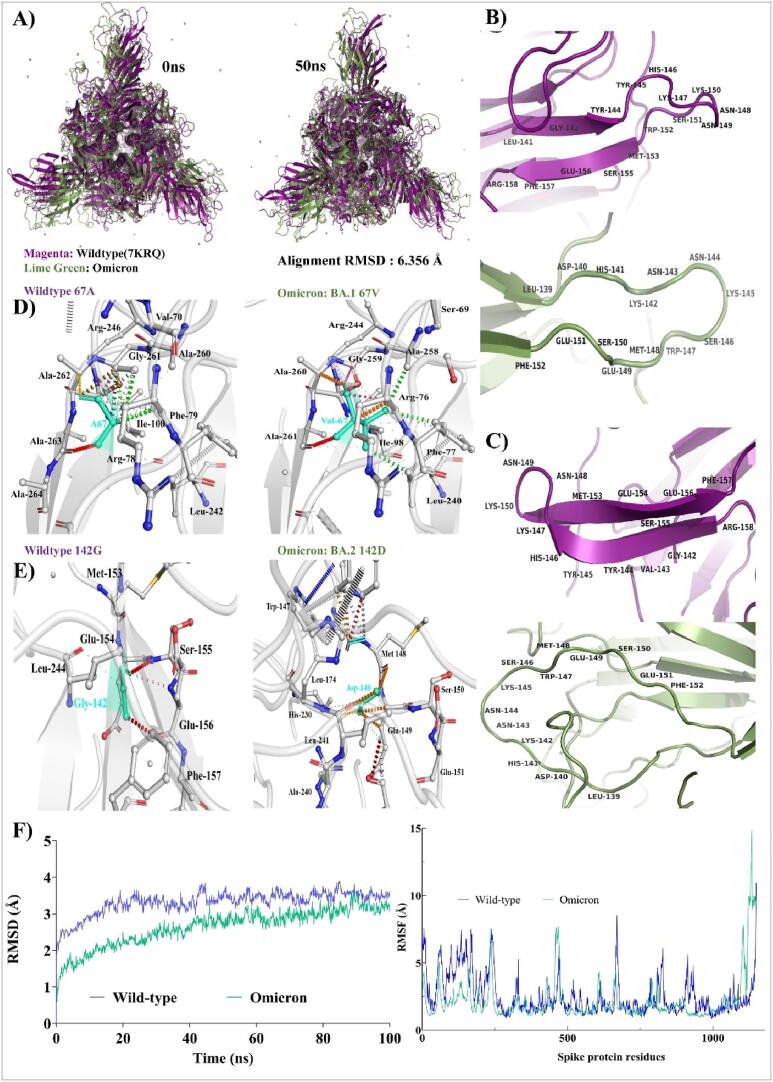
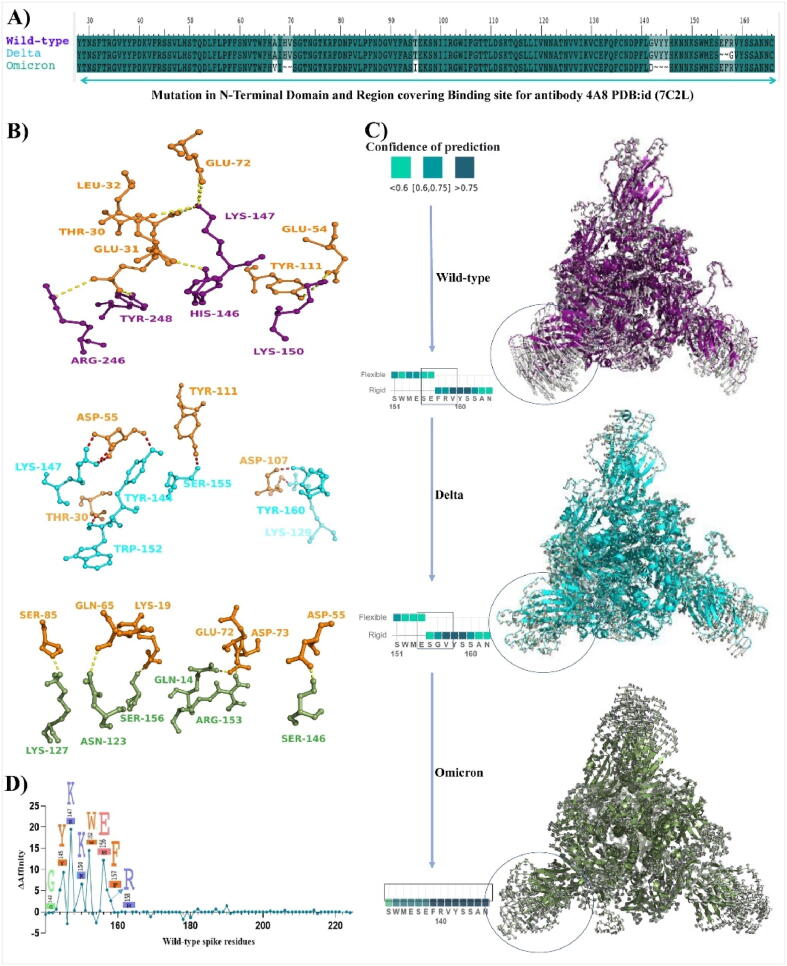
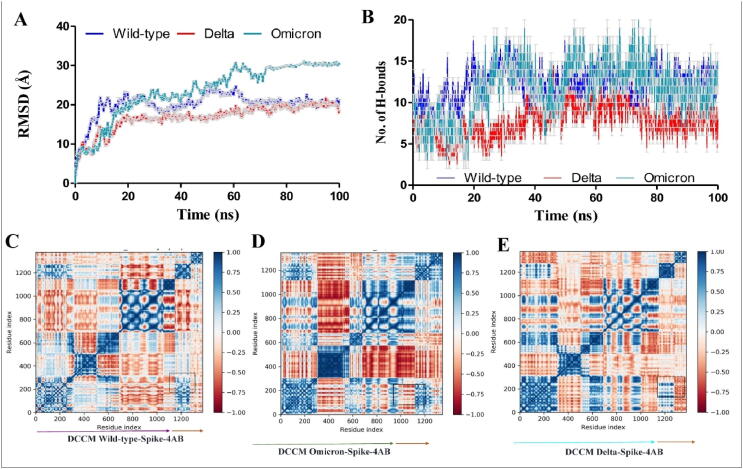
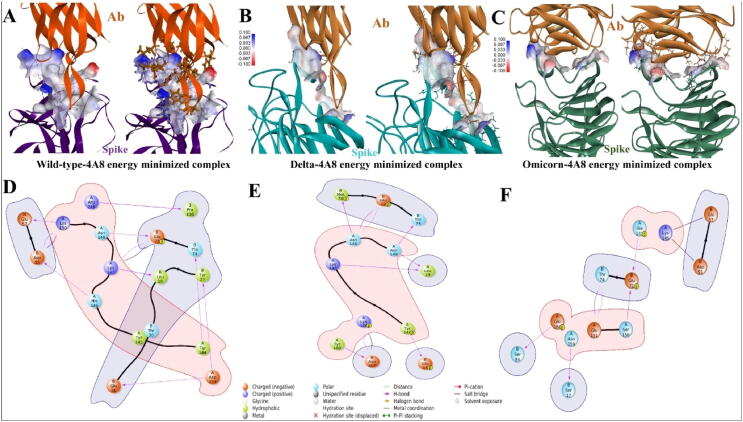
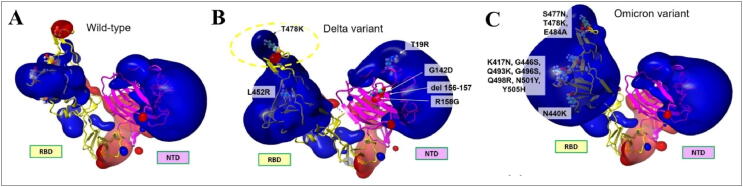
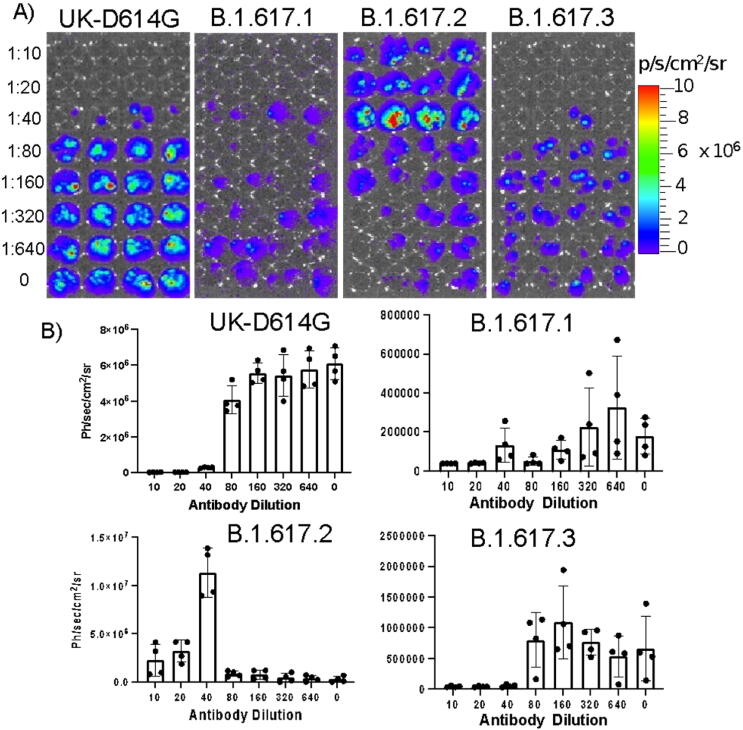
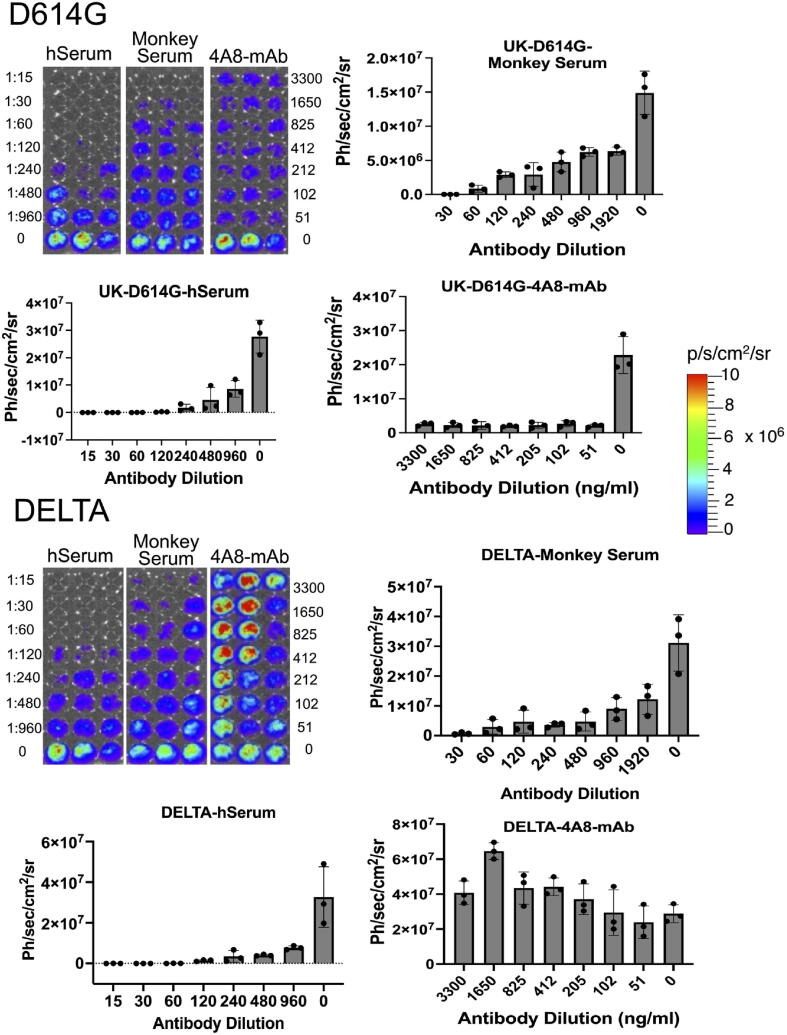
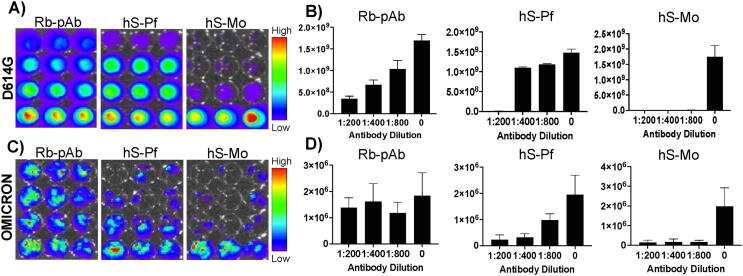
Emerging SARS-CoV-2 variants with higher transmissibility and immune escape remain a persistent threat across the globe. This is evident from the recent outbreaks of the Delta (B.1.617.2) and Omicron variants. These variants have originated from different continents and spread across the globe. In this study, we explored the genomic and structural basis of these variants for their lineage defining mutations of the spike protein through computational analysis, protein modeling, and molecular dynamic (MD) simulations. We further experimentally validated the importance of these deletion mutants for their immune escape using a pseudovirus-based neutralization assay, and an antibody (4A8) that binds directly to the spike protein's NTD. Delta variant with the deletion and mutations in the NTD revealed a better rigidity and reduced flexibility as compared to the wild-type spike protein (Wuhan isolate). Furthermore, computational studies of 4A8 monoclonal antibody (mAb) revealed a reduced binding of Delta variant compared to the wild-type strain. Similarly, the MD simulation data and virus neutralization assays revealed that the Omicron also exhibits immune escape, as antigenic beta-sheets appear to be disrupted. The results of the present study demonstrate the higher possibility of immune escape and thereby achieved better fitness advantages by the Delta and Omicron variants, which warrants further demonstrations through experimental evidences. Our study, based on in-silico computational modelling, simulations, and pseudovirus-based neutralization assay, highlighted and identified the probable mechanism through which the Delta and Omicron variants are more pathogenically evolved with higher transmissibility as compared to the wild-type strain.
Keywords: COVID-19; Immune evasion; In-silico; Pseudovirus; SARS-CoV-2; Spike protein.
© 2022 The Authors.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
Similar articles
-
Emerging Variants of SARS-CoV-2 and Novel Therapeutics Against Coronavirus (COVID-19).2023 May 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2023 May 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 34033342 Free Books & Documents.
-
Wild-type SARS-CoV-2 neutralizing immunity decreases across variants and over time but correlates well with diagnostic testing.Front Immunol. 2023 Feb 8;14:1055429. doi: 10.3389/fimmu.2023.1055429. eCollection 2023. Front Immunol. 2023. PMID: 36845123 Free PMC article.
-
Developing Pseudovirus-Based Neutralization Assay against Omicron-Included SARS-CoV-2 Variants.Viruses. 2022 Jun 18;14(6):1332. doi: 10.3390/v14061332. Viruses. 2022. PMID: 35746803 Free PMC article.
-
The Biological Functions and Clinical Significance of SARS-CoV-2 Variants of Corcern.Front Med (Lausanne). 2022 May 20;9:849217. doi: 10.3389/fmed.2022.849217. eCollection 2022. Front Med (Lausanne). 2022. PMID: 35669924 Free PMC article. Review.
-
Antibody evasion associated with the RBD significant mutations in several emerging SARS-CoV-2 variants and its subvariants.Drug Resist Updat. 2023 Nov;71:101008. doi: 10.1016/j.drup.2023.101008. Epub 2023 Sep 22. Drug Resist Updat. 2023. PMID: 37757651 Review.
Cited by
-
Predictors of COVID-19 Severity in Elderly Patients Infected by Omicron in China, 18 December 2022-5 February 2023.Infect Drug Resist. 2023 Jul 11;16:4505-4518. doi: 10.2147/IDR.S418622. eCollection 2023. Infect Drug Resist. 2023. PMID: 37457796 Free PMC article.
-
Neutralizing and Enhancing Epitopes of the SARS-CoV-2 Receptor-Binding Domain (RBD) Identified by Nanobodies.Viruses. 2023 May 26;15(6):1252. doi: 10.3390/v15061252. Viruses. 2023. PMID: 37376552 Free PMC article.
-
Genomic Surveillance and Molecular Characterization of SARS-CoV-2 Variants During the Peak of the Pandemic in Türkiye.Biochem Genet. 2024 Nov 8. doi: 10.1007/s10528-024-10962-8. Online ahead of print. Biochem Genet. 2024. PMID: 39516327
-
In depth sequencing of a serially sampled household cohort reveals the within-host dynamics of Omicron SARS-CoV-2 and rare selection of novel spike variants.bioRxiv [Preprint]. 2024 Nov 22:2024.11.21.624722. doi: 10.1101/2024.11.21.624722. bioRxiv. 2024. Update in: PLoS Pathog. 2025 Apr 28;21(4):e1013134. doi: 10.1371/journal.ppat.1013134. PMID: 39605326 Free PMC article. Updated. Preprint.
-
T Cell Peptide Prediction, Immune Response, and Host-Pathogen Relationship in Vaccinated and Recovered from Mild COVID-19 Subjects.Biomolecules. 2024 Sep 26;14(10):1217. doi: 10.3390/biom14101217. Biomolecules. 2024. PMID: 39456150 Free PMC article.
References
-
- Ai, J., Wang, X., He, X., Zhao, X., Zhang, Y., Jiang, Y., Li, M., Cui, Y., Chen, Y., Qiao, R., Li, L., Yang, L., Li, Y., Hu, Z., Zhang, W., & Wang, P. (2022). Antibody evasion of SARS-CoV-2 Omicron BA.1, BA.1.1, BA.2, and BA.3 sub-lineages. Cell host & microbe, S1931-3128(22)00243-8. Advance online publication. https://doi.org/10.1016/j.chom.2022.05.001. - PMC - PubMed
-
- Bayani F., Safaei Hashkavaei N., Uversky V.N., Mozaffari-Jovin S., Sefidbakht Y. Insights into the structural peculiarities of the N-terminal and receptor binding domains of the spike protein from the SARS-CoV-2 Omicron variant. Comput Biol Med. 2022;147 doi: 10.1016/j.compbiomed.2022.105735. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous