

CRISPR/Cas9-based genome-wide screening for deubiquitinase subfamily identifies USP1 regulating MAST1-driven cisplatin-resistance in cancer cells
- PMID: 35966591
- PMCID: PMC9373805
- DOI: 10.7150/thno.72826
CRISPR/Cas9-based genome-wide screening for deubiquitinase subfamily identifies USP1 regulating MAST1-driven cisplatin-resistance in cancer cells
Abstract
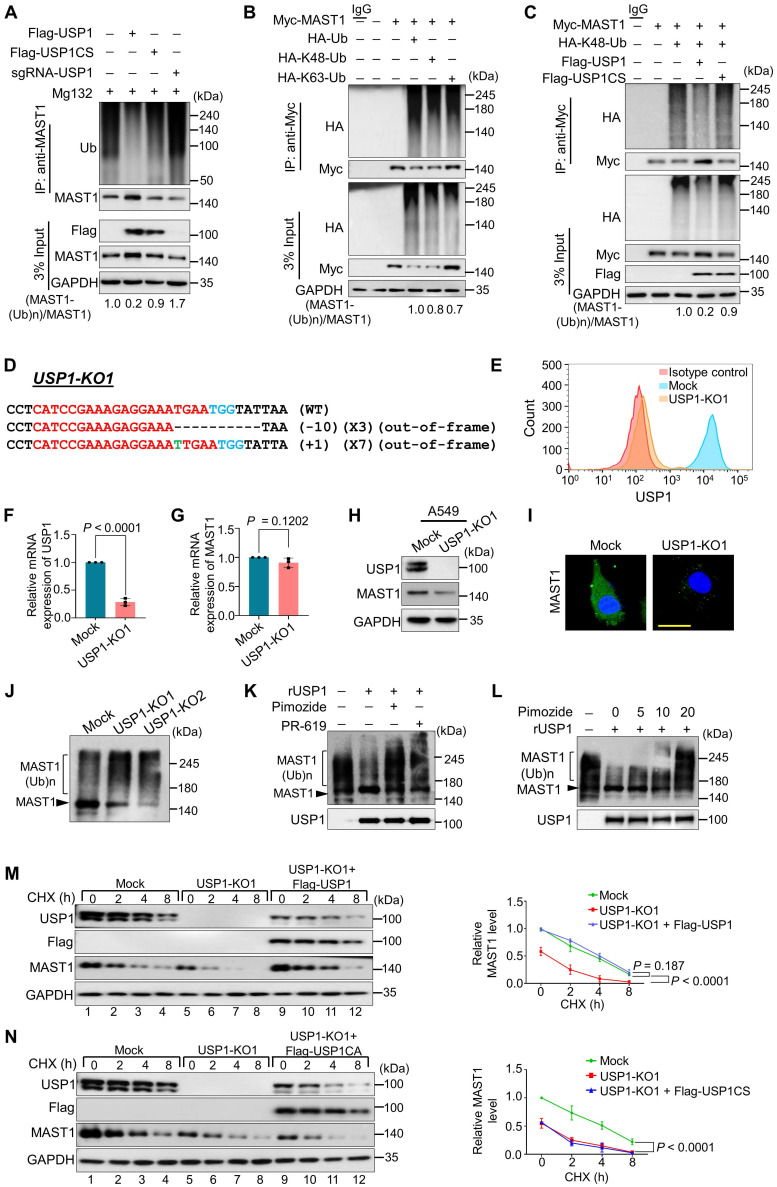
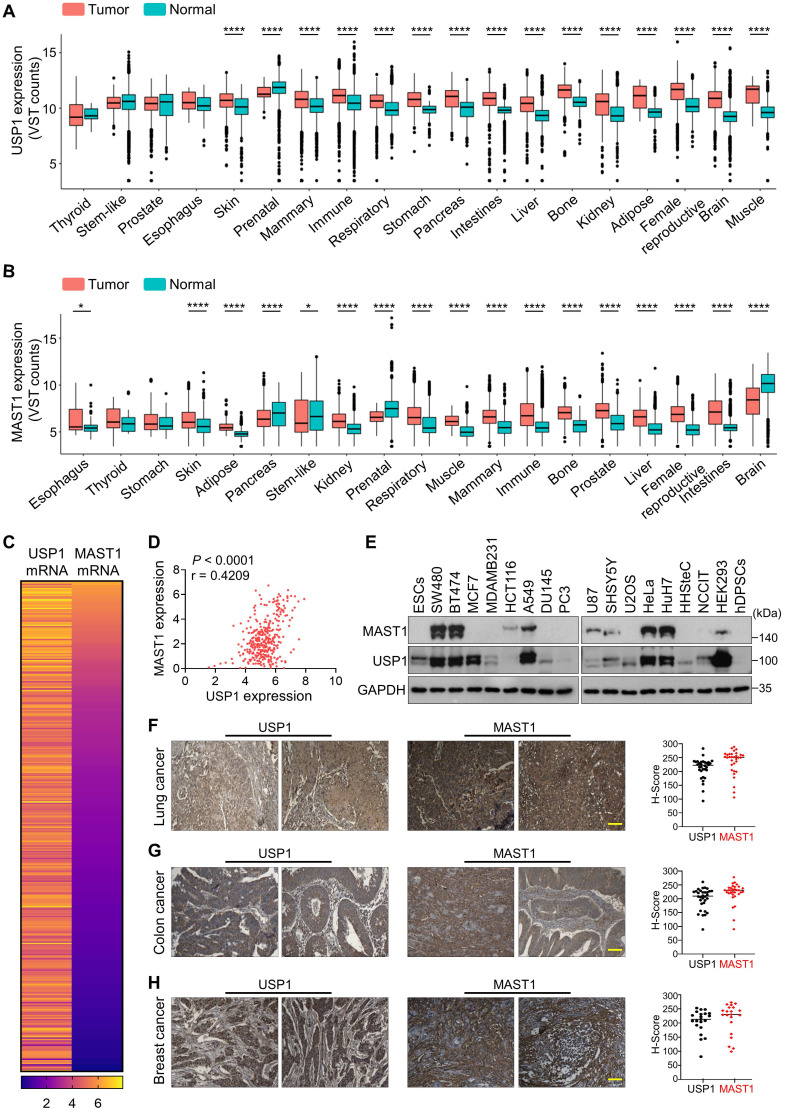
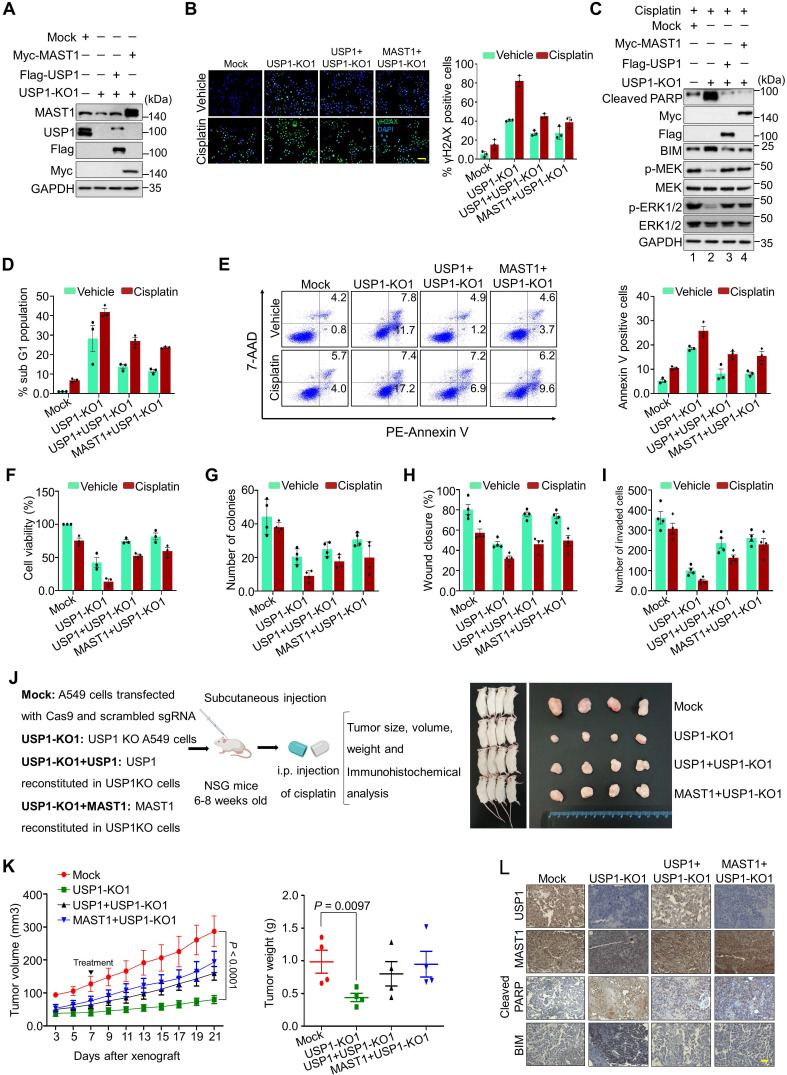
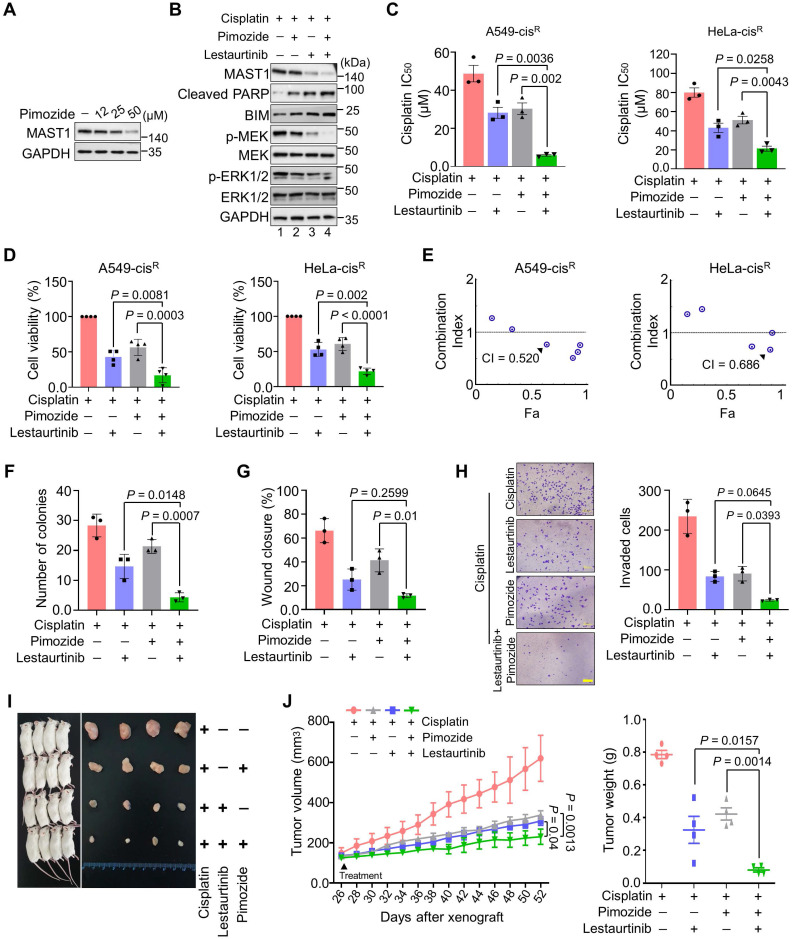
Background: Cisplatin is one of the frontline anticancer agents. However, development of cisplatin-resistance limits the therapeutic efficacy of cisplatin-based treatment. The expression of microtubule-associated serine/threonine kinase 1 (MAST1) is a primary factor driving cisplatin-resistance in cancers by rewiring the MEK pathway. However, the mechanisms responsible for MAST1 regulation in conferring drug resistance is unknown. Methods: We implemented a CRISPR/Cas9-based, genome-wide, dual screening system to identify deubiquitinating enzymes (DUBs) that govern cisplatin resistance and regulate MAST1 protein level. We analyzed K48- and K63-linked polyubiquitination of MAST1 protein and mapped the interacting domain between USP1 and MAST1 by immunoprecipitation assay. The deubiquitinating effect of USP1 on MAST1 protein was validated using rescue experiments, in vitro deubiquitination assay, immunoprecipitation assays, and half-life analysis. Furthermore, USP1-knockout A549 lung cancer cells were generated to validate the deubiquitinating activity of USP1 on MAST1 abundance. The USP1-MAST1 correlation was evaluated using bioinformatics tool and in different human clinical tissues. The potential role of USP1 in regulating MAST1-mediated cisplatin resistance was confirmed using a series of in vitro and in vivo experiments. Finally, the clinical relevance of the USP1-MAST1 axis was validated by application of small-molecule inhibitors in a lung cancer xenograft model in NSG mice. Results: The CRISPR/Cas9-based dual screening system identified USP1 as a novel deubiquitinase that interacts, stabilizes, and extends the half-life of MAST1 by preventing its K48-linked polyubiquitination. The expression analysis across human clinical tissues revealed a positive correlation between USP1 and MAST1. USP1 promotes MAST1-mediated MEK1 activation as an underlying mechanism that contributes to cisplatin-resistance in cancers. Loss of USP1 led to attenuation of MAST1-mediated cisplatin-resistance both in vitro and in vivo. The combined pharmacological inhibition of USP1 and MAST1 using small-molecule inhibitors further abrogated MAST1 level and synergistically enhanced cisplatin efficacy in a mouse xenograft model. Conclusions: Overall, our study highlights the role of USP1 in the development of cisplatin resistance and uncovers the regulatory mechanism of MAST1-mediated cisplatin resistance in cancers. Co-treatment with USP1 and MAST1 inhibitors abrogated tumor growth and synergistically enhanced cisplatin efficacy, suggesting a novel alternative combinatorial therapeutic strategy that could further improve MAST1-based therapy in patients with cisplatin-resistant tumors.
Keywords: Apoptosis; DNA damage; DUB inhibitor; clinical tumor samples; drug resistance; kinase inhibitor; ubiquitin proteasome system.
© The author(s).
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
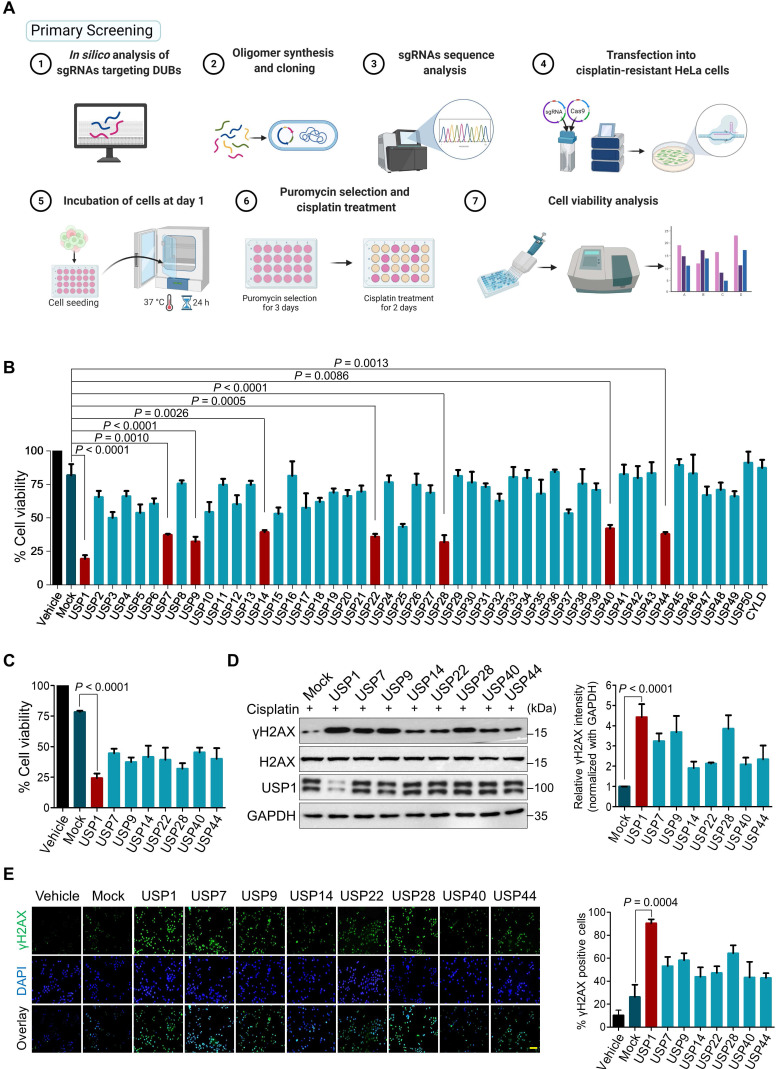
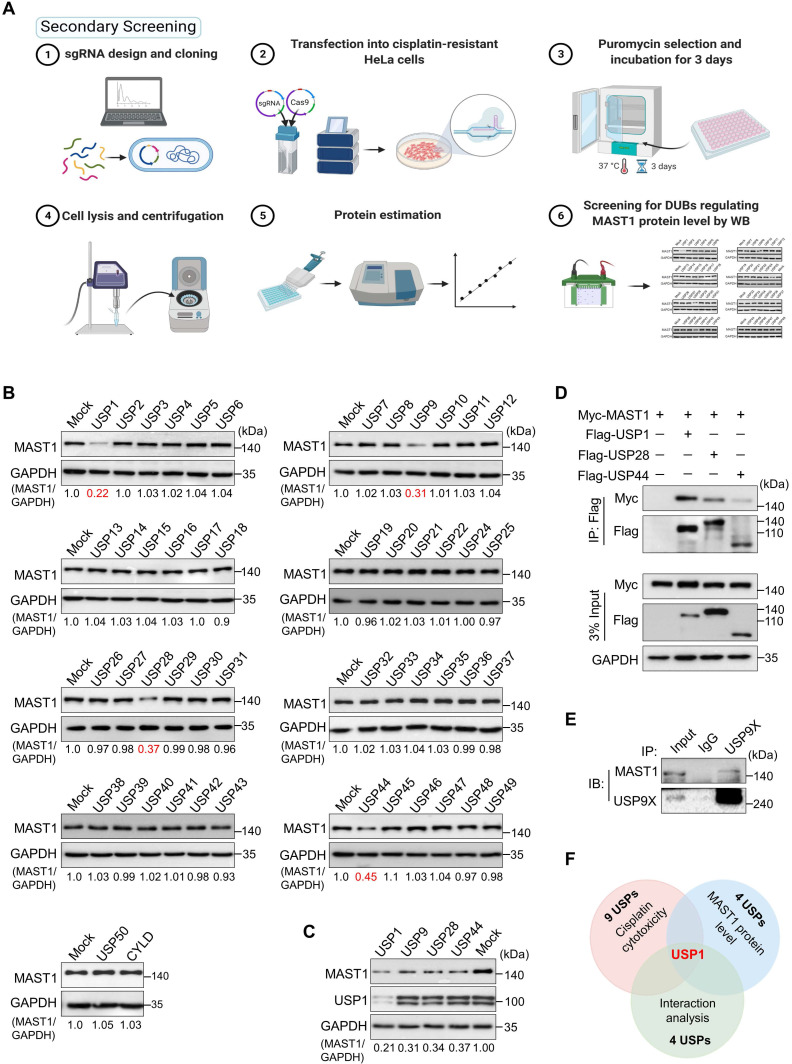
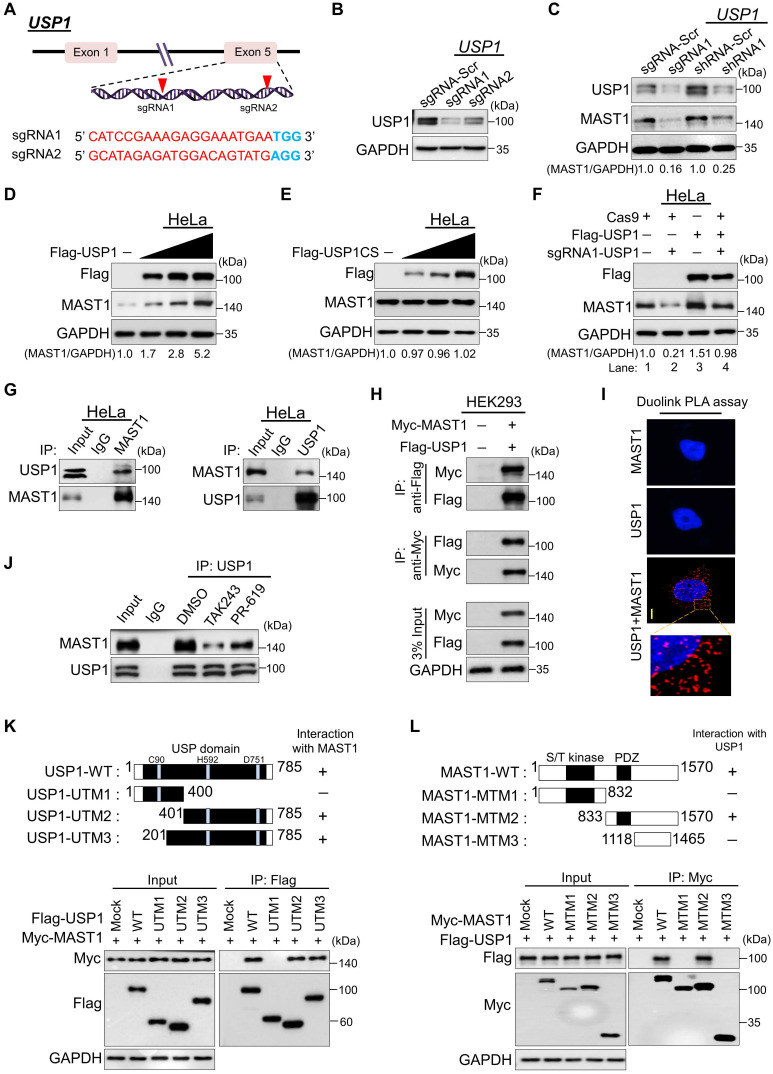
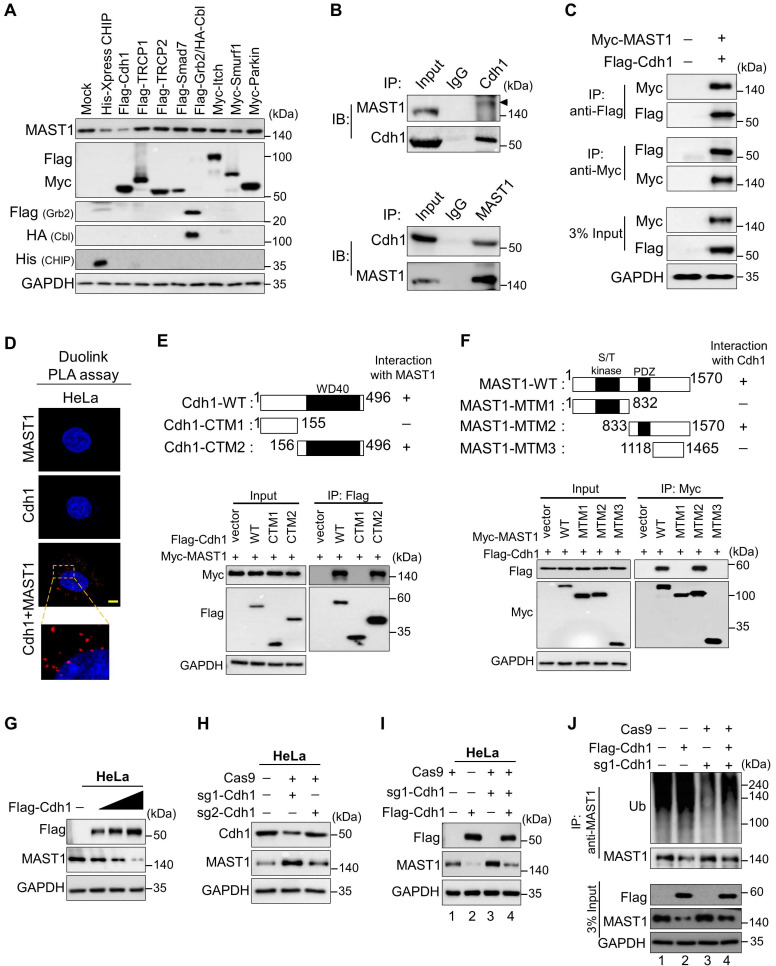
References
-
- Galanski M. Recent developments in the field of anticancer platinum complexes. Recent patents on anti-cancer drug discovery. 2006;1:285–95. - PubMed
-
- Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–79. - PubMed
-
- Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O. et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–83. - PubMed
-
- Wang J, Zhou JY, Wu GS. ERK-dependent MKP-1-mediated cisplatin resistance in human ovarian cancer cells. Cancer research. 2007;67:11933–41. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
