

The dynamic transcriptome during maturation of biofilms formed by methicillin-resistant Staphylococcus aureus
- PMID: 35966712
- PMCID: PMC9366926
- DOI: 10.3389/fmicb.2022.882346
The dynamic transcriptome during maturation of biofilms formed by methicillin-resistant Staphylococcus aureus
Abstract
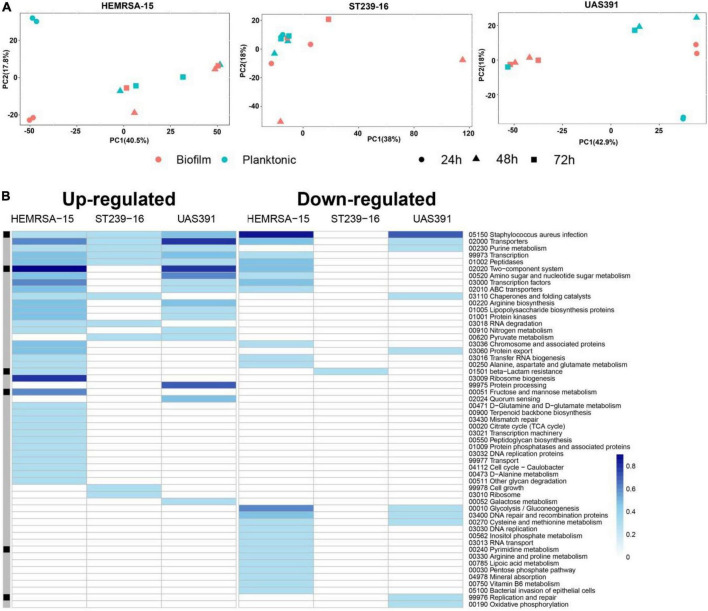
Background: Methicillin-resistant Staphylococcus aureus (MRSA), a leading cause of chronic infections, forms prolific biofilms which afford an escape route from antibiotic treatment and host immunity. However, MRSA clones are genetically diverse, and mechanisms underlying biofilm formation remain under-studied. Such studies form the basis for developing targeted therapeutics. Here, we studied the temporal changes in the biofilm transcriptome of three pandemic MRSA clones: USA300, HEMRSA-15, and ST239.
Methods: Biofilm formation was assessed using a static model with one representative strain per clone. Total RNA was extracted from biofilm and planktonic cultures after 24, 48, and 72 h of growth, followed by rRNA depletion and sequencing (Illumina Inc., San Diego, CA, United States, NextSeq500, v2, 1 × 75 bp). Differentially expressed gene (DEG) analysis between phenotypes and among early (24 h), intermediate (48 h), and late (72 h) stages of biofilms was performed together with in silico co-expression network construction and compared between clones. To understand the influence of SCCmec and ACME on biofilm formation, isogenic mutants containing deletions of the entire elements or of single genes therein were constructed in USA300.
Results: Genes involved in primarily core genome-encoded KEGG pathways (transporters and others) were upregulated in 24-h biofilm culture compared to 24-h planktonic culture. However, the number of affected pathways in the ST239 24 h biofilm (n = 11) was remarkably lower than that in USA300/EMRSA-15 biofilms (USA300: n = 27, HEMRSA-15: n = 58). The clfA gene, which encodes clumping factor A, was the single common DEG identified across the three clones in 24-h biofilm culture (2.2- to 2.66-fold). In intermediate (48 h) and late (72 h) stages of biofilms, decreased expression of central metabolic and fermentative pathways (glycolysis/gluconeogenesis, fatty acid biosynthesis), indicating a shift to anaerobic conditions, was already evident in USA300 and HEMRSA-15 in 48-h biofilm cultures; ST239 showed a similar profile at 72 h. Last, SCCmec+ACME deletion and opp3D disruption negatively affected USA300 biofilm formation.
Conclusion: Our data show striking differences in gene expression during biofilm formation by three of the most important pandemic MRSA clones, USA300, HEMRSA-15, and ST239. The clfA gene was the only significantly upregulated gene across all three strains in 24-h biofilm cultures and exemplifies an important target to disrupt early biofilms. Furthermore, our data indicate a critical role for arginine catabolism pathways in early biofilm formation.
Keywords: MRSA; Staphylococcus aureus; biofilm; clumping factor A; transcriptome.
Copyright © 2022 Vlaeminck, Lin, Xavier, De Backer, Berkell, De Greve, Hernalsteens, Kumar-Singh, Goossens and Malhotra-Kumar.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

References
-
- Atshan S. S., Shamsudin M. N., Karunanidhi A., van Belkum A., Lung L. T. T., Sekawi Z., et al. (2013). Quantitative PCR analysis of genes expressed during biofilm development of methicillin resistant Staphylococcus aureus (MRSA). Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 18 106–112. 10.1016/j.meegid.2013.05.002 - DOI - PubMed
-
- Bastian M., Heymann S., Jacomy M. (2009). Gephi: an open source software for exploring and manipulating networks. Proc. Int. AAAI Conf. Web Soc. Media 3 361–362.
LinkOut - more resources
Full Text Sources