

Efficient derivation of chimeric-antigen receptor-modified TSCM cells
- PMID: 35967430
- PMCID: PMC9366550
- DOI: 10.3389/fimmu.2022.877682
Efficient derivation of chimeric-antigen receptor-modified TSCM cells
Abstract
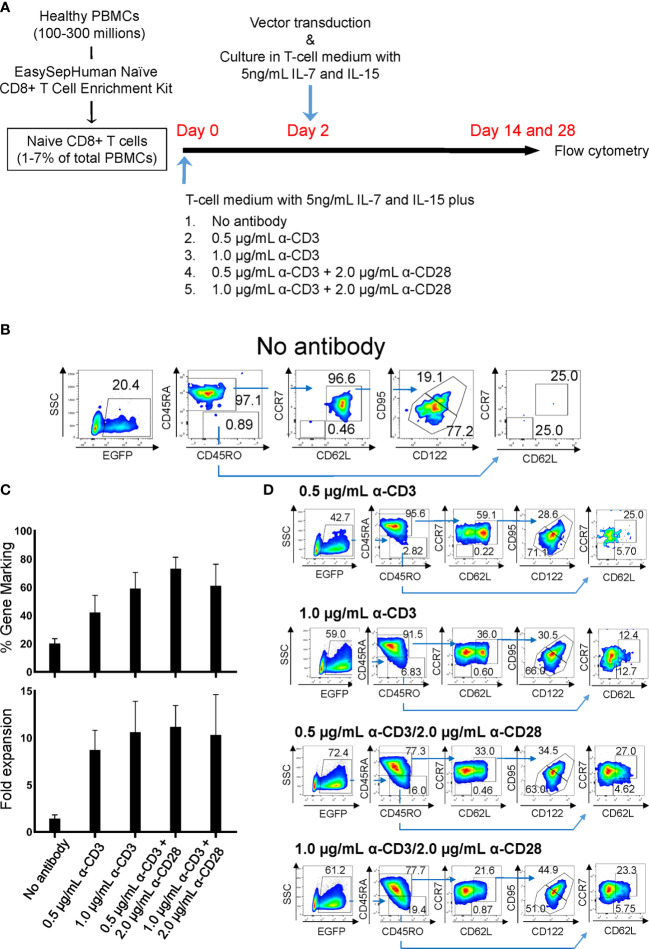
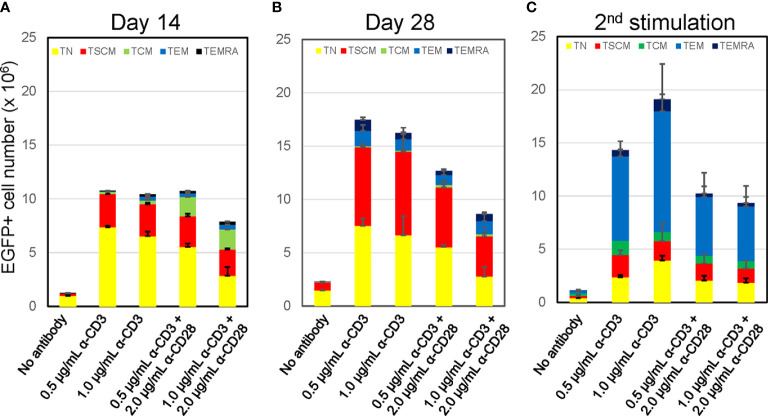
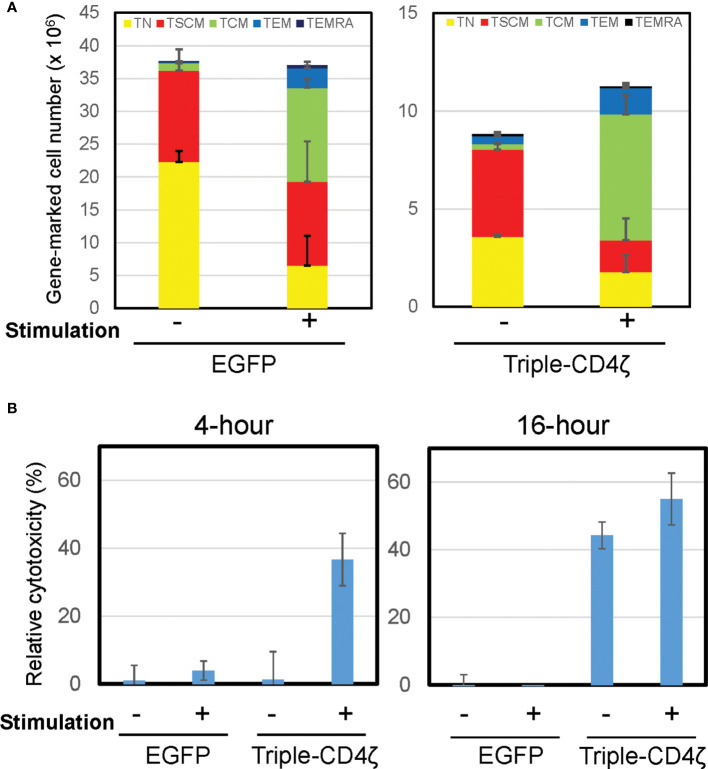
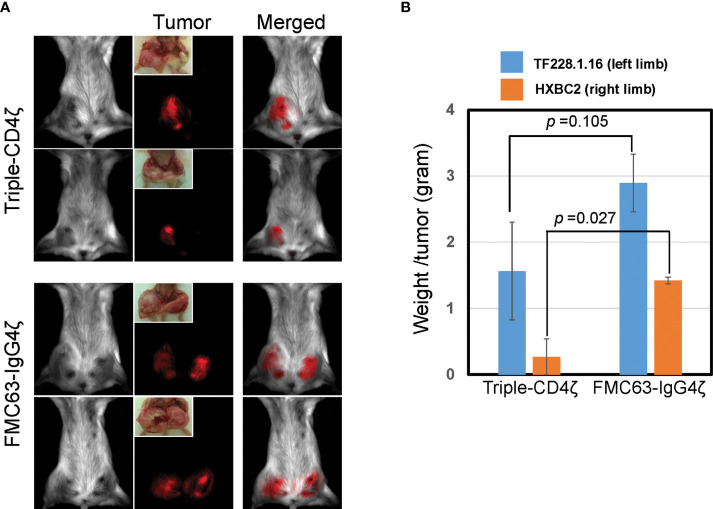
Chimeric-antigen receptor (CAR) T-cell immunotherapy employs autologous-T cells modified with an antigen-specific CAR. Current CAR-T manufacturing processes tend to yield products dominated by effector T cells and relatively small proportions of long-lived memory T cells. Those few cells are a so-called stem cell memory T (TSCM) subset, which express naïve T-cell markers and are capable of self-renewal and oligopotent differentiation into effector phenotypes. Increasing the proportion of this subset may lead to more effective therapies by improving CAR-T persistence; however, there is currently no standardized protocol for the effective generation of CAR-TSCM cells. Here we present a simplified protocol enabling efficient derivation of gene-modified TSCM cells: Stimulation of naïve CD8+ T cells with only soluble anti-CD3 antibody and culture with IL-7 and IL-15 was sufficient for derivation of CD8+ T cells harboring TSCM phenotypes and oligopotent capabilities. These in-vitro expanded TSCM cells were engineered with CARs targeting the HIV-1 envelope protein as well as the CD19 molecule and demonstrated effector activity both in vitro and in a xenograft mouse model. This simple protocol for the derivation of CAR-TSCM cells may facilitate improved adoptive immunotherapy.
Keywords: CAR; HIV-1; TSCM; adoptive immunotherapy; gene therapy.
Copyright © 2022 Kranz, Kuhlmann, Chan, Kim, Chen and Kamata.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Induction of a central memory and stem cell memory phenotype in functionally active CD4+ and CD8+ CAR T cells produced in an automated good manufacturing practice system for the treatment of CD19+ acute lymphoblastic leukemia.Cancer Immunol Immunother. 2018 Jul;67(7):1053-1066. doi: 10.1007/s00262-018-2155-7. Epub 2018 Mar 31. Cancer Immunol Immunother. 2018. PMID: 29605883 Free PMC article.
-
Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies.Blood. 2016 Jul 28;128(4):519-28. doi: 10.1182/blood-2015-11-683847. Epub 2016 May 25. Blood. 2016. PMID: 27226436 Free PMC article.
-
Next-Generation Manufacturing Protocols Enriching TSCM CAR T Cells Can Overcome Disease-Specific T Cell Defects in Cancer Patients.Front Immunol. 2020 Jun 19;11:1217. doi: 10.3389/fimmu.2020.01217. eCollection 2020. Front Immunol. 2020. PMID: 32636841 Free PMC article.
-
Generation of CAR-TSCM: CAR-T with super clutch.Int Immunopharmacol. 2024 Jul 30;136:112379. doi: 10.1016/j.intimp.2024.112379. Epub 2024 Jun 3. Int Immunopharmacol. 2024. PMID: 38833844 Review.
-
Advances in modular control of CAR-T therapy with adapter-mediated CARs.Adv Drug Deliv Rev. 2022 Aug;187:114358. doi: 10.1016/j.addr.2022.114358. Epub 2022 May 23. Adv Drug Deliv Rev. 2022. PMID: 35618140 Free PMC article. Review.
Cited by
-
CAR-T cell therapy for hematological malignancies: Limitations and optimization strategies.Front Immunol. 2022 Sep 28;13:1019115. doi: 10.3389/fimmu.2022.1019115. eCollection 2022. Front Immunol. 2022. PMID: 36248810 Free PMC article. Review.
-
Memory stem CD8+T cells in HIV/Mtb mono- and co-infection: characteristics, implications, and clinical significance.Front Cell Infect Microbiol. 2024 Dec 10;14:1485825. doi: 10.3389/fcimb.2024.1485825. eCollection 2024. Front Cell Infect Microbiol. 2024. PMID: 39720790 Free PMC article. Review.
-
Recent advances on anti-HIV chimeric antigen receptor-T-cell treatment to provide sustained HIV remission.Curr Opin HIV AIDS. 2024 Jul 1;19(4):169-178. doi: 10.1097/COH.0000000000000858. Epub 2024 May 1. Curr Opin HIV AIDS. 2024. PMID: 38695148 Free PMC article. Review.
-
CCR5 gene editing and HIV immunotherapy: current understandings, challenges, and future directions.Front Immunol. 2025 Jun 18;16:1590690. doi: 10.3389/fimmu.2025.1590690. eCollection 2025. Front Immunol. 2025. PMID: 40607431 Free PMC article.
-
Extracellular domain, hinge, and transmembrane determinants affecting surface CD4 expression of a novel anti-HIV chimeric antigen receptor (CAR) construct.PLoS One. 2024 Aug 12;19(8):e0293990. doi: 10.1371/journal.pone.0293990. eCollection 2024. PLoS One. 2024. PMID: 39133676 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials