

A comprehensive in silico exploration of the impacts of missense variants on two different conformations of human pirin protein
- PMID: 35967515
- PMCID: PMC9362109
- DOI: 10.1186/s42269-022-00917-7
A comprehensive in silico exploration of the impacts of missense variants on two different conformations of human pirin protein
Abstract
Background: Pirin, a member of the cupin superfamily, is an iron-binding non-heme protein. It acts as a coregulator of several transcription factors, especially the members of NFκB transcription factor family. Based on the redox state of its iron cofactor, it can assume two different conformations and thereby act as a redox sensor inside the nucleus. Previous studies suggested that pirin may be associated with cancer, inflammatory diseases as well as COVID-19 severities. Hence, it is important to explore the pathogenicity of its missense variants. In this study, we used a number of in silico tools to investigate the effects of missense variants of pirin on its structure, stability, metal cofactor binding affinity and interactions with partner proteins. In addition, we used protein dynamics simulation to elucidate the effects of selected variants on its dynamics. Furthermore, we calculated the frequencies of haplotypes containing pirin missense variants across five major super-populations (African, Admixed American, East Asian, European and South Asian).
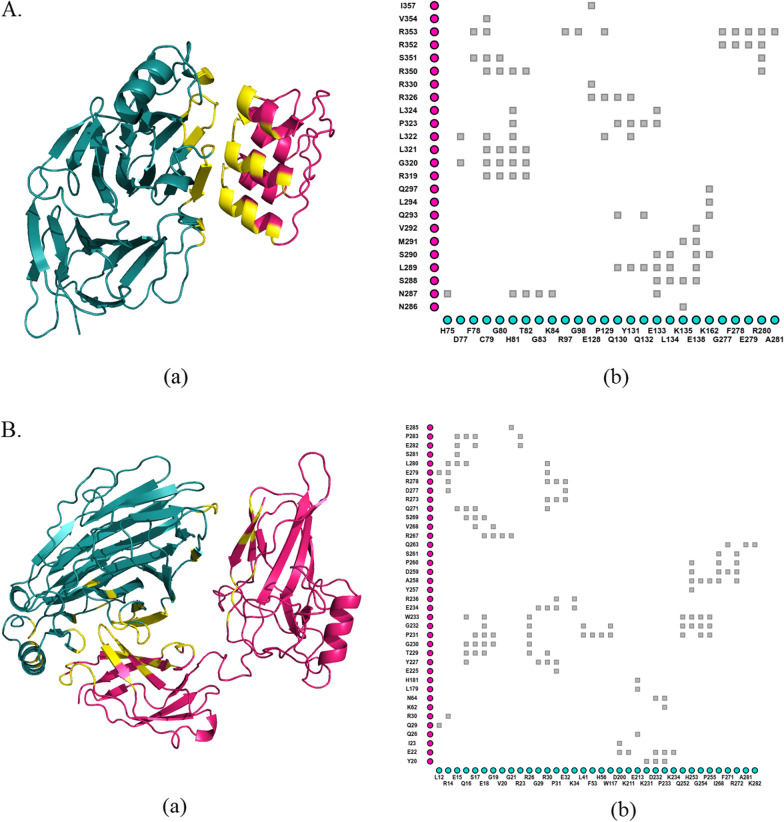
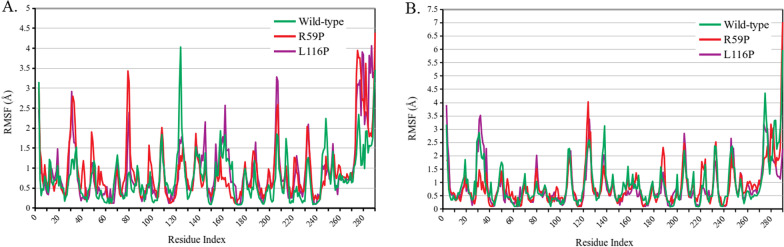
Results: Among a total of 153 missense variants of pirin, 45 were uniformly predicted to be pathogenic. Of these, seven variants can be considered for further experimental studies. Variants R59P and L116P were predicted to significantly destabilize and damage pirin structure, substantially reduce its affinity to its binding partners and alter pirin residue fluctuation profile via changing the flexibility of several key residues. Additionally, variants R59Q, F78V, G98D, V151D and L220P were found to impact pirin structure and function in multiple ways. As no haplotype was identified to be harboring more than one missense variant, further interrogation of the individual effects of these seven missense variants is highly recommended.
Conclusions: Pirin is involved in the transcriptional regulation of several genes and can play an important role in inflammatory responses. The variants predicted to be pathogenic in this study may thus contribute to a better understanding of the underlying molecular mechanisms of various inflammatory diseases. Future studies should be focused on clarifying if any of these variants can be used as disease biomarkers.
Supplementary information: The online version contains supplementary material available at 10.1186/s42269-022-00917-7.
Keywords: Cancer; Inflammation; NFκB pathway; Non-heme protein; Oxidative stress; Pathogenic variants; Pirin; Transcriptional regulation.
© The Author(s) 2022.
Conflict of interest statement
Competing interestsThe authors declare that they have no competing interests.
Figures
Similar articles
-
Role of the redox state of the Pirin-bound cofactor on interaction with the master regulators of inflammation and other pathways.PLoS One. 2023 Nov 30;18(11):e0289158. doi: 10.1371/journal.pone.0289158. eCollection 2023. PLoS One. 2023. PMID: 38033031 Free PMC article.
-
Pirin is an iron-dependent redox regulator of NF-κB.Proc Natl Acad Sci U S A. 2013 Jun 11;110(24):9722-7. doi: 10.1073/pnas.1221743110. Epub 2013 May 28. Proc Natl Acad Sci U S A. 2013. PMID: 23716661 Free PMC article.
-
Crystal structure of human pirin: an iron-binding nuclear protein and transcription cofactor.J Biol Chem. 2004 Jan 9;279(2):1491-8. doi: 10.1074/jbc.M310022200. Epub 2003 Oct 22. J Biol Chem. 2004. PMID: 14573596
-
Role of Pirin, an Oxidative Stress Sensor Protein, in Epithelial Carcinogenesis.Biology (Basel). 2021 Feb 4;10(2):116. doi: 10.3390/biology10020116. Biology (Basel). 2021. PMID: 33557375 Free PMC article. Review.
-
Analysis and Interpretation of the Impact of Missense Variants in Cancer.Int J Mol Sci. 2021 May 21;22(11):5416. doi: 10.3390/ijms22115416. Int J Mol Sci. 2021. PMID: 34063805 Free PMC article. Review.
References
-
- Betts MJ, Russell RB. Amino acid properties and consequences of substitutions. Bioinform Genet. 2003;317:10–1002.
LinkOut - more resources
Full Text Sources
Research Materials