

Cobamide Sharing Is Predicted in the Human Skin Microbiome
- PMID: 35968974
- PMCID: PMC9600381
- DOI: 10.1128/msystems.00677-22
Cobamide Sharing Is Predicted in the Human Skin Microbiome
Abstract
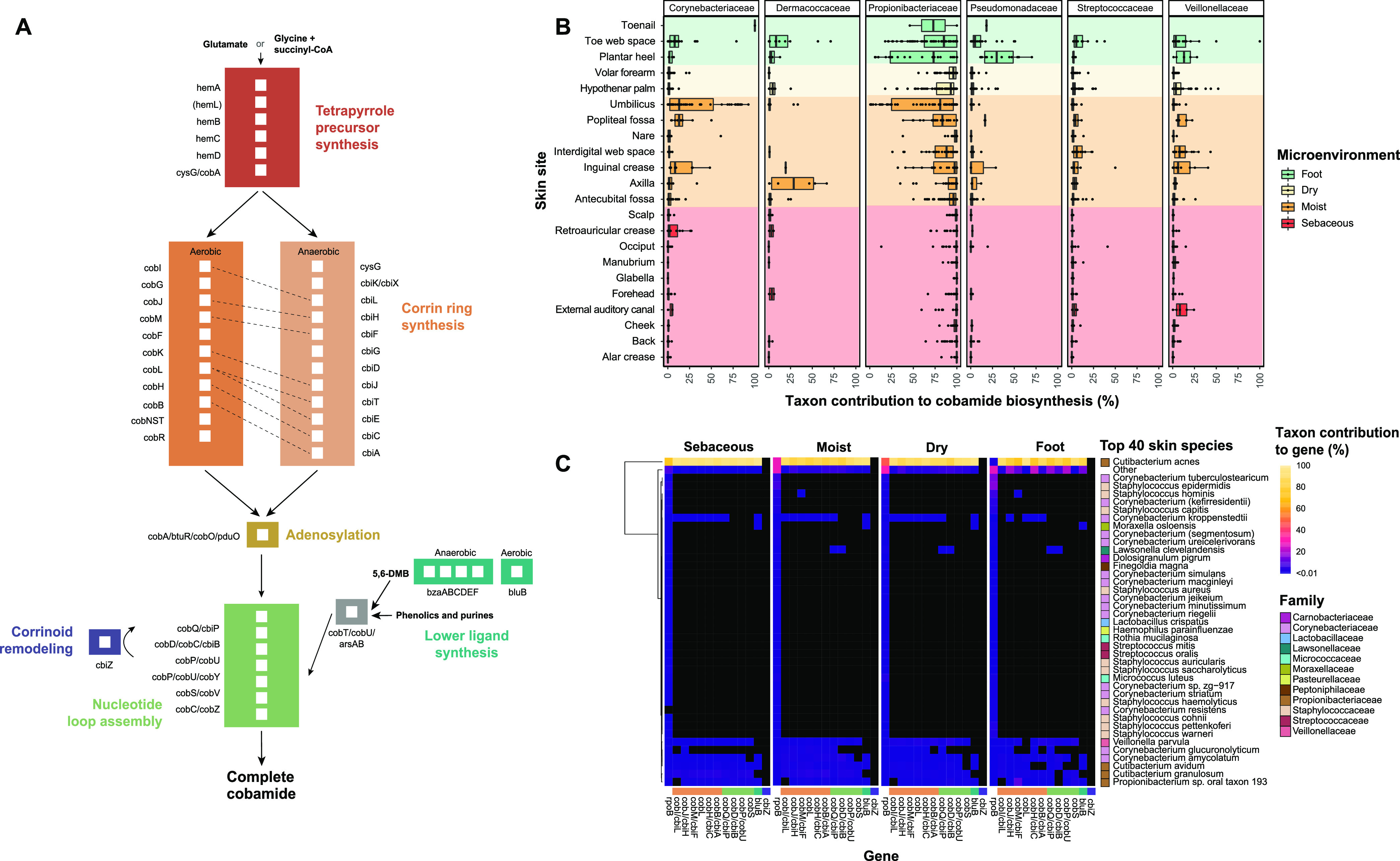
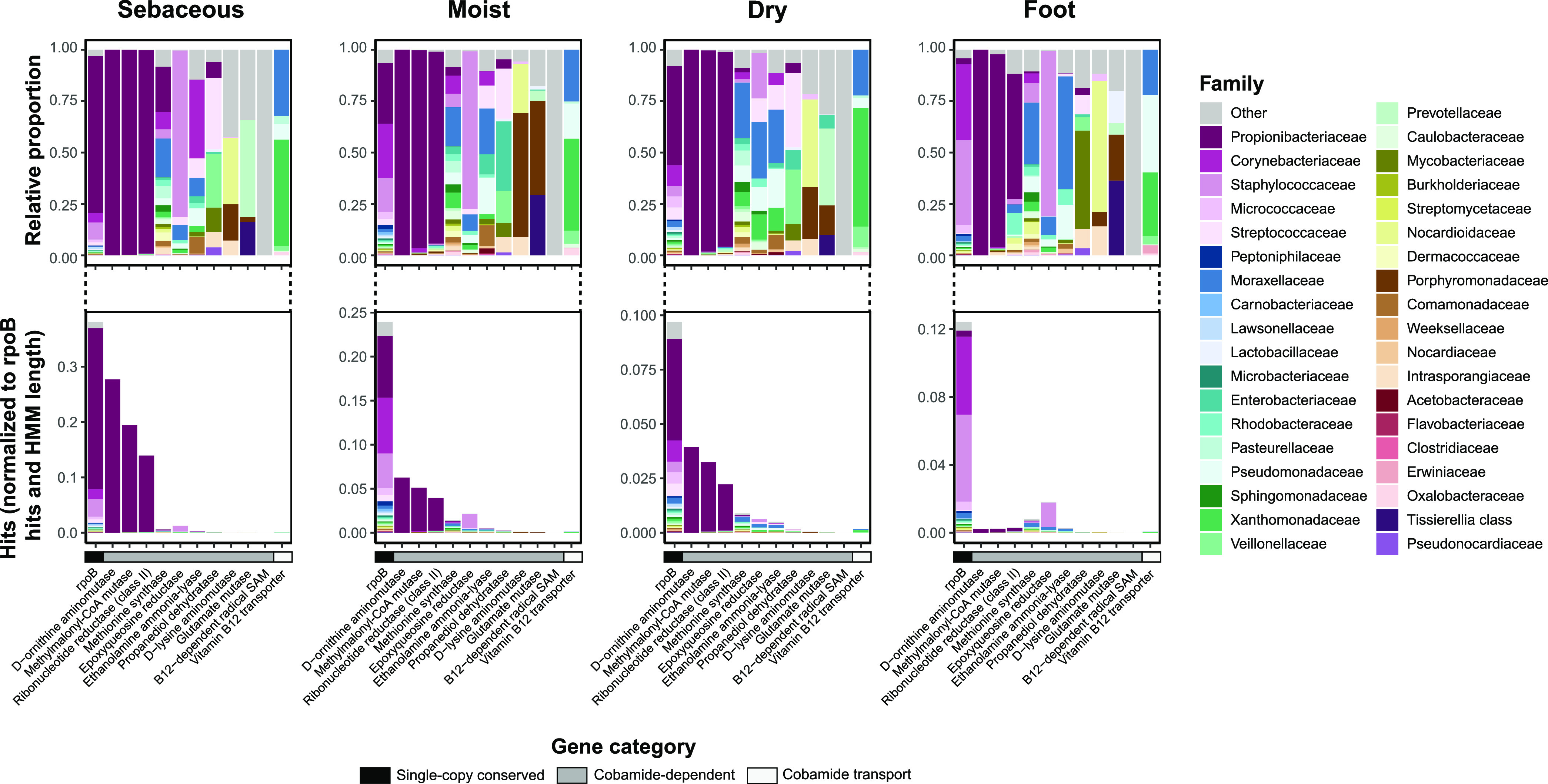
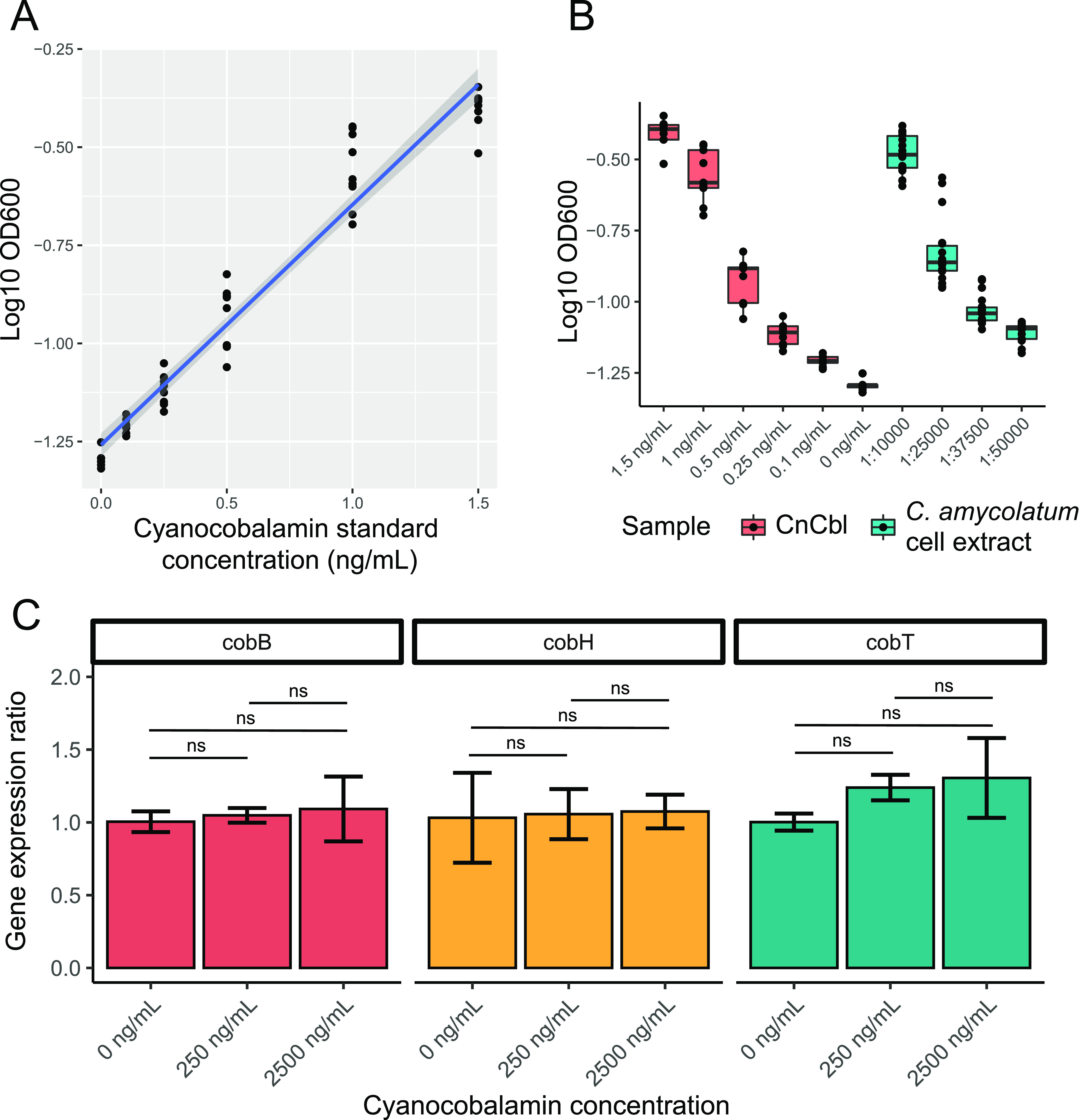
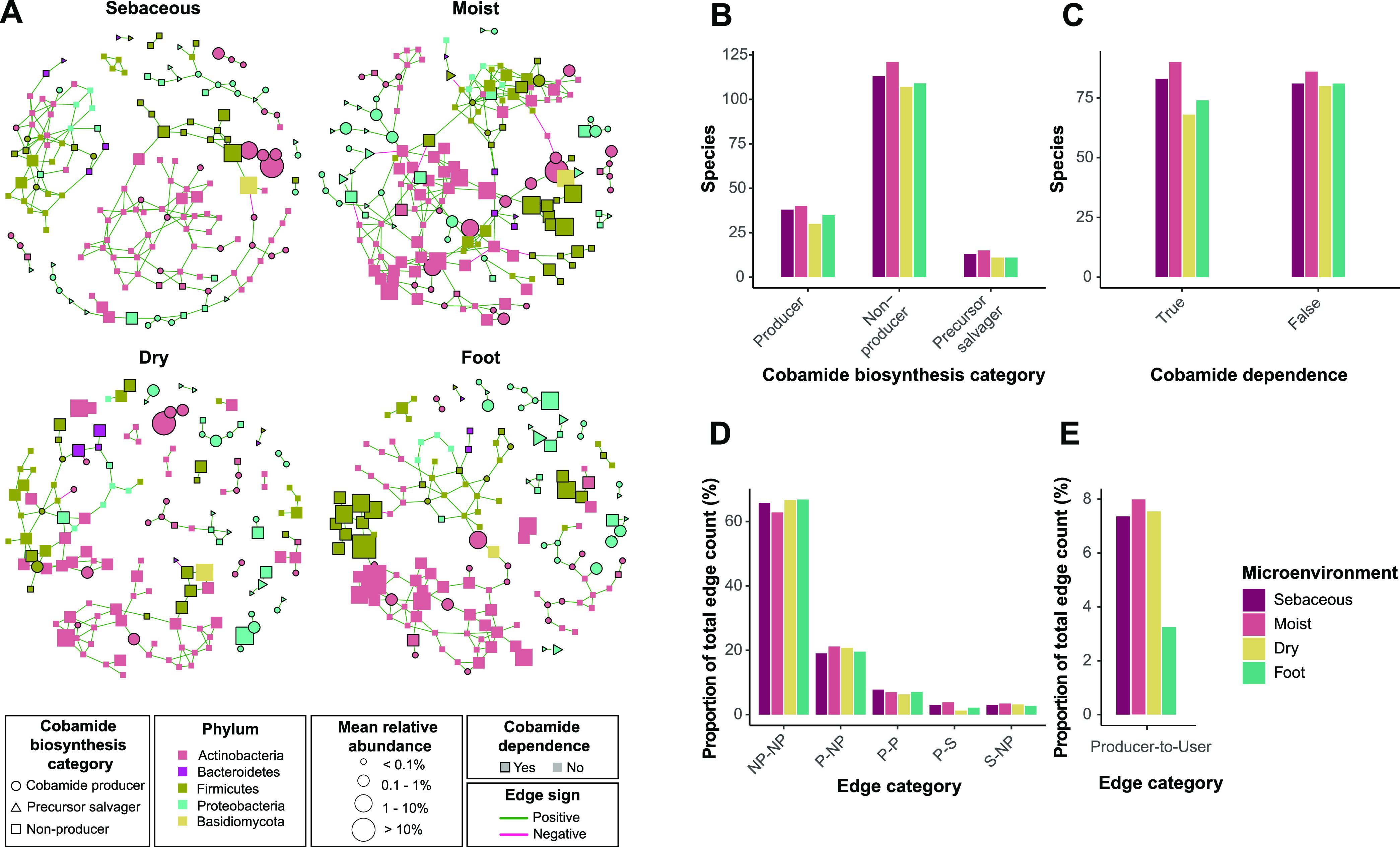
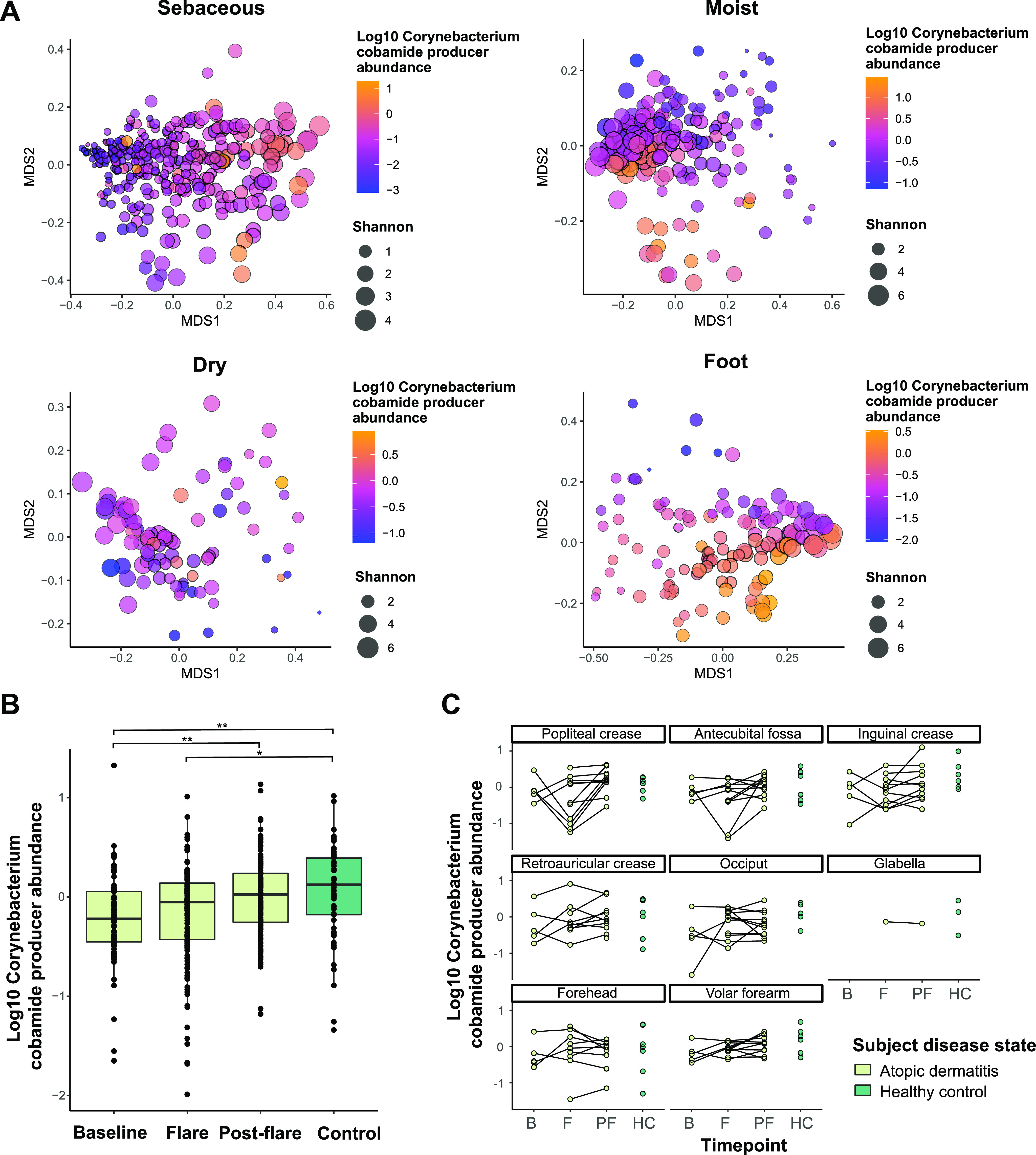
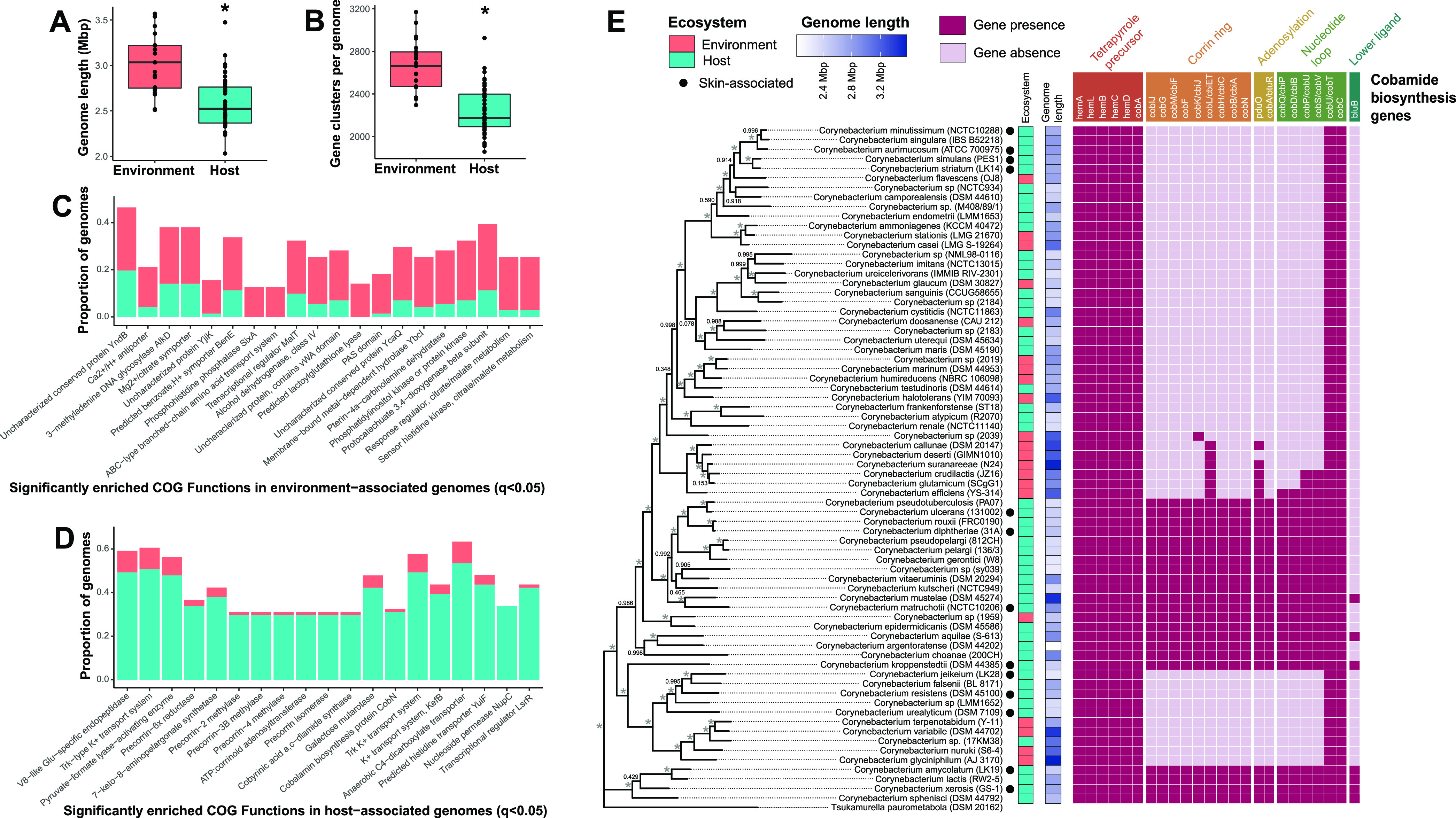
The skin microbiome is a key player in human health, with diverse functions ranging from defense against pathogens to education of the immune system. While recent studies have begun to shed light on the valuable role that skin microorganisms have in maintaining the skin barrier, a detailed understanding of the complex interactions that shape healthy skin microbial communities is limited. Cobamides, the vitamin B12 class of cofactor, are essential for organisms across the tree of life. Because this vitamin is only produced by a limited fraction of prokaryotes, cobamide sharing is predicted to mediate community dynamics within microbial communities. Here, we provide the first large-scale metagenomic assessment of cobamide biosynthesis and utilization in the skin microbiome. We show that while numerous and diverse taxa across the major bacterial phyla on the skin encode cobamide-dependent enzymes, relatively few species encode de novo cobamide biosynthesis. We show that cobamide producers and users are integrated into the network structure of microbial communities across the different microenvironments of the skin and that changes in microbiome community structure and diversity are associated with the abundance of cobamide producers in the Corynebacterium genus, for both healthy and diseased skin states. Finally, we find that de novo cobamide biosynthesis is enriched only in Corynebacterium species associated with hosts, including those prevalent on human skin. We confirm that the cofactor is produced in excess through quantification of cobamide production by human skin-associated species isolated in the laboratory. Taken together, our results reveal the potential for cobamide sharing within skin microbial communities, which we hypothesize mediates microbiome community dynamics and host interactions. IMPORTANCE The skin microbiome is essential for maintaining skin health and function. However, the microbial interactions that dictate microbiome structure, stability, and function are not well understood. Here, we investigate the biosynthesis and use of cobamides, a cofactor needed by many organisms but only produced by select prokaryotes, within the human skin microbiome. We found that while a large proportion of skin taxa encode cobamide-dependent enzymes, only a select few encode de novo cobamide biosynthesis. Further, the abundance of cobamide-producing Corynebacterium species is associated with skin microbiome diversity and structure, and within this genus, de novo biosynthesis is enriched in host-associated species compared to environment-associated species. These findings identify cobamides as a potential mediator of skin microbiome dynamics and skin health.
Keywords: cobamides; ecology; genomics; metagenomics; skin microbiome.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Skin-associated Corynebacterium amycolatum shares cobamides.bioRxiv [Preprint]. 2024 Apr 28:2024.04.28.591522. doi: 10.1101/2024.04.28.591522. bioRxiv. 2024. Update in: mSphere. 2025 Jan 28;10(1):e0060624. doi: 10.1128/msphere.00606-24. PMID: 38712214 Free PMC article. Updated. Preprint.
-
Skin-associated Corynebacterium amycolatum shares cobamides.mSphere. 2025 Jan 28;10(1):e0060624. doi: 10.1128/msphere.00606-24. Epub 2024 Dec 18. mSphere. 2025. PMID: 39692507 Free PMC article.
-
Microbes display broad diversity in cobamide preferences.mSystems. 2025 Apr 22;10(4):e0140724. doi: 10.1128/msystems.01407-24. Epub 2025 Mar 21. mSystems. 2025. PMID: 40116488 Free PMC article.
-
Comparative Analysis of Corrinoid Profiles across Host-Associated and Environmental Samples.Biochemistry. 2022 Dec 20;61(24):2791-2796. doi: 10.1021/acs.biochem.2c00367. Epub 2022 Aug 29. Biochemistry. 2022. PMID: 36037062 Review.
-
Sharing vitamins: Cobamides unveil microbial interactions.Science. 2020 Jul 3;369(6499):eaba0165. doi: 10.1126/science.aba0165. Science. 2020. PMID: 32631870 Free PMC article. Review.
Cited by
-
Recent advances in single-cell engineered live biotherapeutic products research for skin repair and disease treatment.NPJ Biofilms Microbiomes. 2023 Dec 8;9(1):95. doi: 10.1038/s41522-023-00463-8. NPJ Biofilms Microbiomes. 2023. PMID: 38065982 Free PMC article. Review.
-
vRhyme enables binning of viral genomes from metagenomes.Nucleic Acids Res. 2022 Aug 12;50(14):e83. doi: 10.1093/nar/gkac341. Nucleic Acids Res. 2022. PMID: 35544285 Free PMC article.
-
Ankle brachial indices and anaerobes: is peripheral arterial disease associated with anaerobic bacteria in diabetic foot ulcers?Ther Adv Endocrinol Metab. 2022 Aug 23;13:20420188221118747. doi: 10.1177/20420188221118747. eCollection 2022. Ther Adv Endocrinol Metab. 2022. PMID: 36051573 Free PMC article.
-
Skin-associated Corynebacterium amycolatum shares cobamides.bioRxiv [Preprint]. 2024 Apr 28:2024.04.28.591522. doi: 10.1101/2024.04.28.591522. bioRxiv. 2024. Update in: mSphere. 2025 Jan 28;10(1):e0060624. doi: 10.1128/msphere.00606-24. PMID: 38712214 Free PMC article. Updated. Preprint.
-
Skin-associated Corynebacterium amycolatum shares cobamides.mSphere. 2025 Jan 28;10(1):e0060624. doi: 10.1128/msphere.00606-24. Epub 2024 Dec 18. mSphere. 2025. PMID: 39692507 Free PMC article.
References
-
- Scharschmidt TC, Vasquez KS, Pauli ML, Leitner EG, Chu K, Truong HA, Lowe MM, Sanchez Rodriguez R, Ali N, Laszik ZG, Sonnenburg JL, Millar SE, Rosenblum MD. 2017. Commensal microbes and hair follicle morphogenesis coordinately drive Treg migration into neonatal skin. Cell Host Microbe 21:467–477.e5. doi:10.1016/j.chom.2017.03.001. - DOI - PMC - PubMed
-
- Constantinides MG, Link VM, Tamoutounour S, Wong AC, Perez-Chaparro PJ, Han S-J, Chen YE, Li K, Farhat S, Weckel A, Krishnamurthy SR, Vujkovic-Cvijin I, Linehan JL, Bouladoux N, Merrill ED, Roy S, Cua DJ, Adams EJ, Bhandoola A, Scharschmidt TC, Aubé J, Fischbach MA, Belkaid Y. 2019. MAIT cells are imprinted by the microbiota in early life and promote tissue repair. Science 366:eaax6624. doi:10.1126/science.aax6624. - DOI - PMC - PubMed
-
- Linehan JL, Harrison OJ, Han S-J, Byrd AL, Vujkovic-Cvijin I, Villarino AV, Sen SK, Shaik J, Smelkinson M, Tamoutounour S, Collins N, Bouladoux N, Dzutsev A, Rosshart SP, Arbuckle JH, Wang C-R, Kristie TM, Rehermann B, Trinchieri G, Brenchley JM, O'Shea JJ, Belkaid Y. 2018. Non-classical immunity controls microbiota impact on skin immunity and tissue Repair. Cell 172:784–796.e18. doi:10.1016/j.cell.2017.12.033. - DOI - PMC - PubMed
-
- Di Domizio J, Belkhodja C, Chenuet P, Fries A, Murray T, Mondéjar PM, Demaria O, Conrad C, Homey B, Werner S, Speiser DE, Ryffel B, Gilliet M. 2020. The commensal skin microbiota triggers type I IFN-dependent innate repair responses in injured skin. Nat Immunol 21:1034–1045. doi:10.1038/s41590-020-0721-6. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R35GM137828/HHS | NIH | National Institute of General Medical Sciences (NIGMS)
- T32AI055397/HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID)
- U19AI142720/HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID)
- U19 AI142720/AI/NIAID NIH HHS/United States
- R35 GM137828/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
