

Single-Cell Dissection of the Multiomic Landscape of High-Grade Serous Ovarian Cancer
- PMID: 35969151
- PMCID: PMC9627134
- DOI: 10.1158/0008-5472.CAN-21-3819
Single-Cell Dissection of the Multiomic Landscape of High-Grade Serous Ovarian Cancer
Abstract
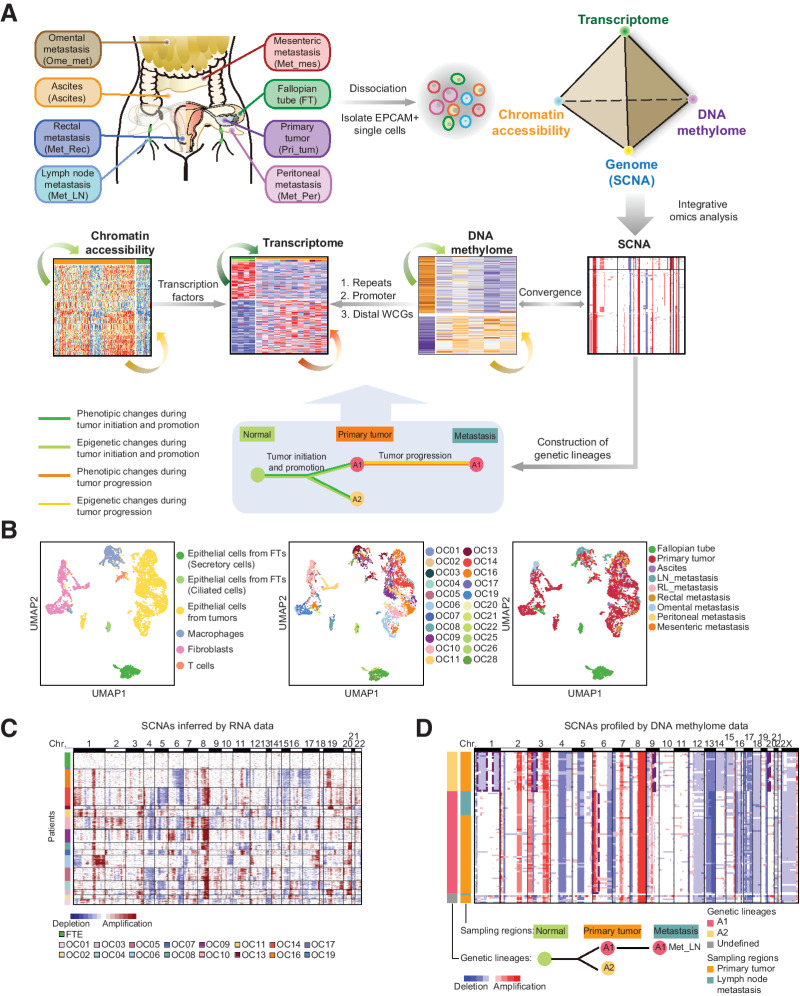
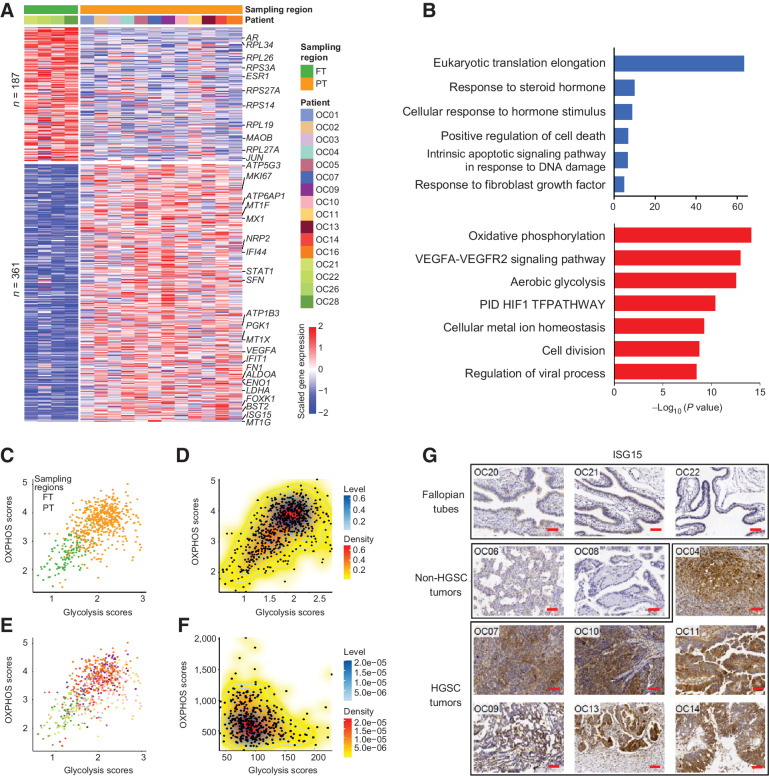
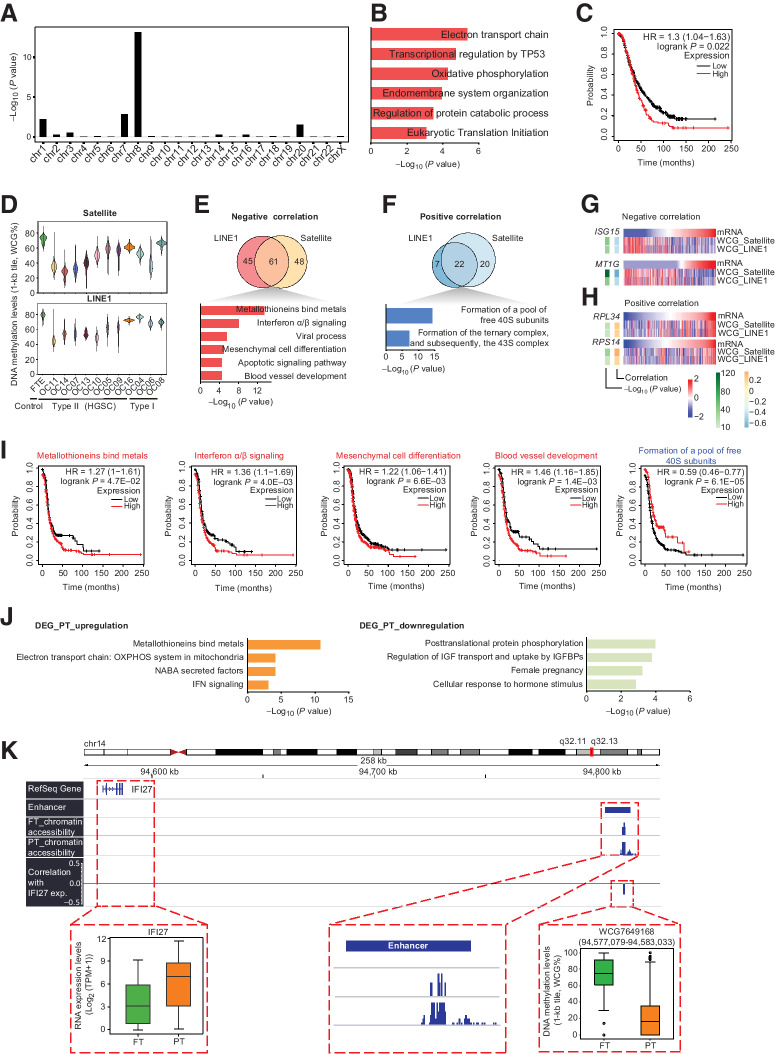
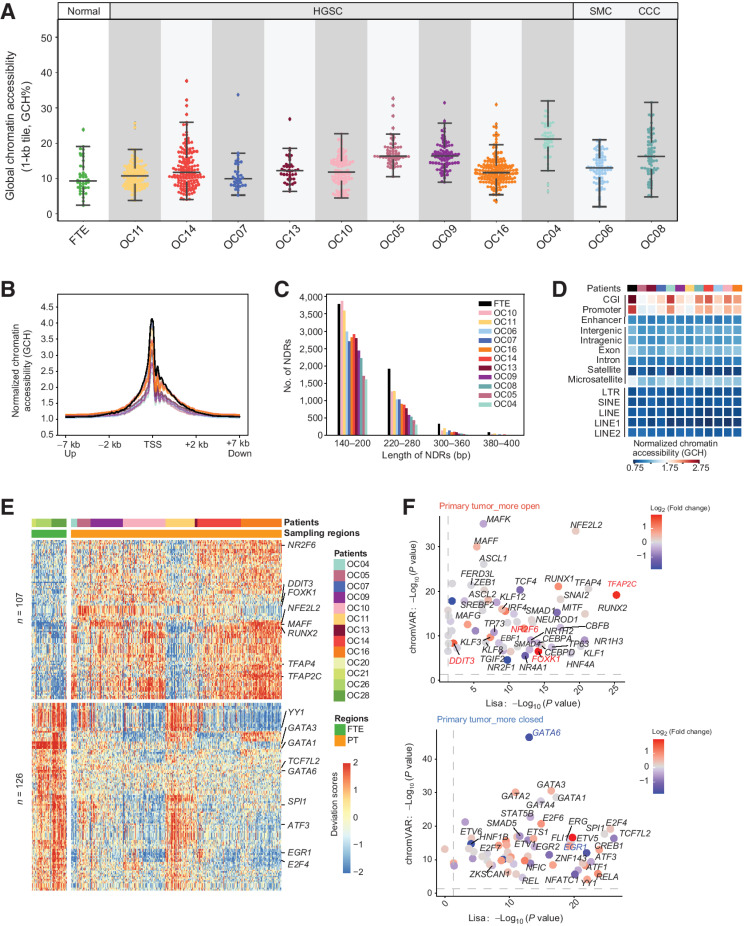
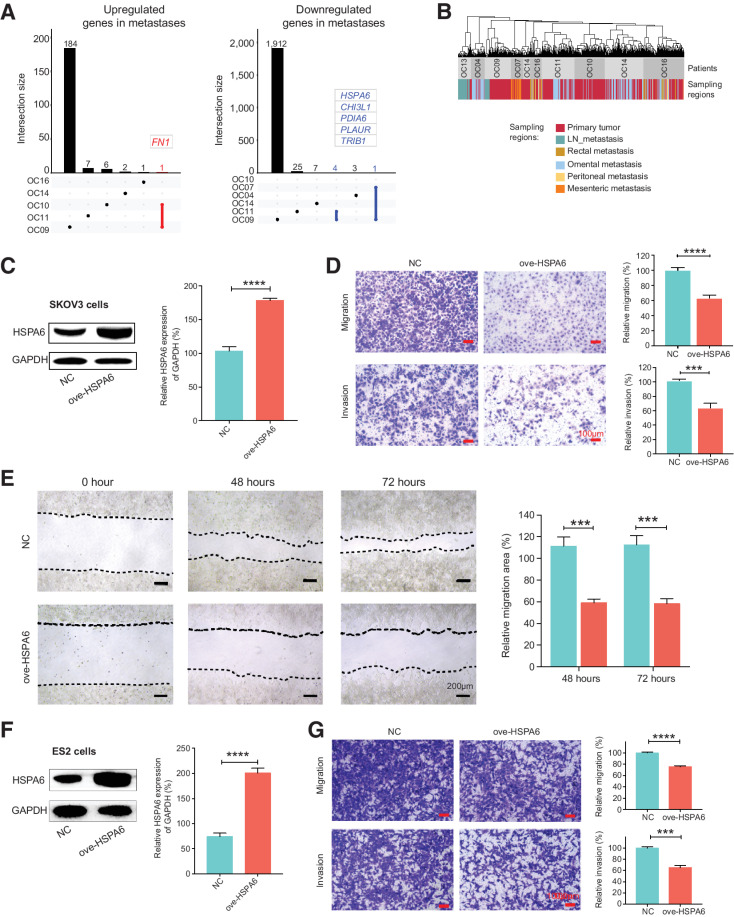
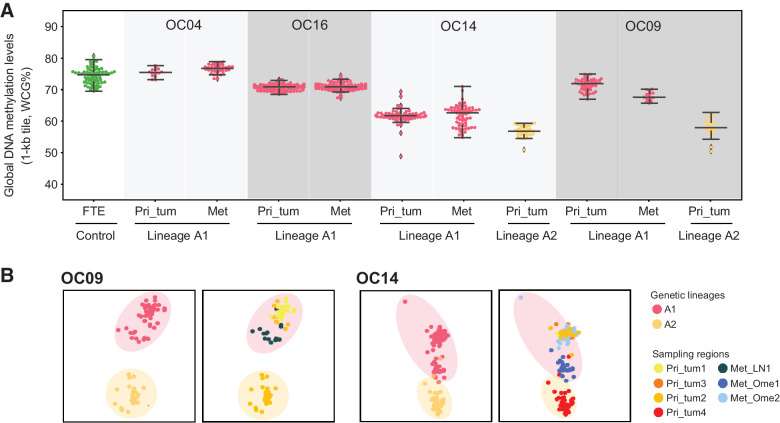
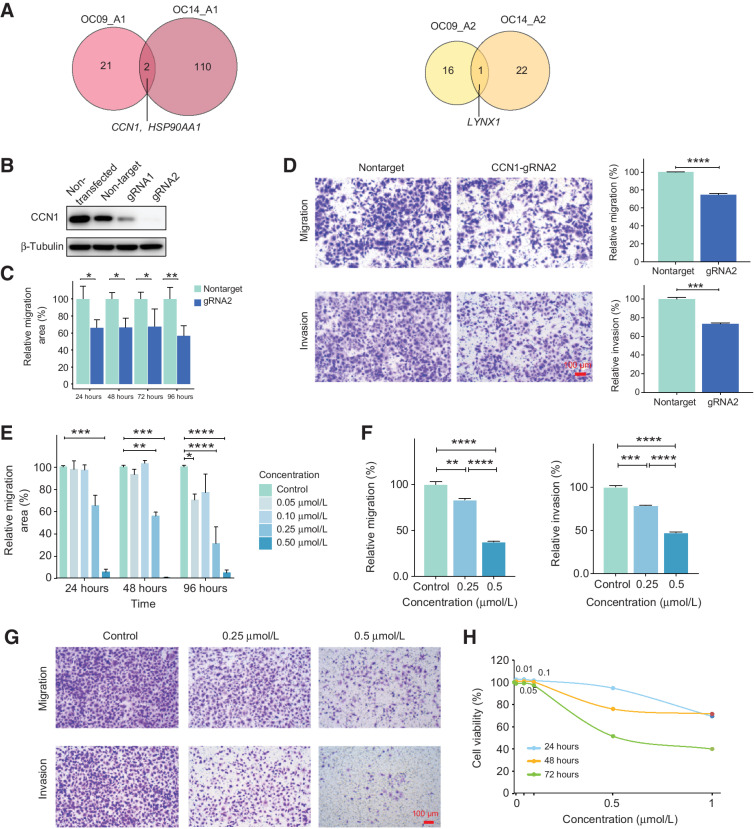
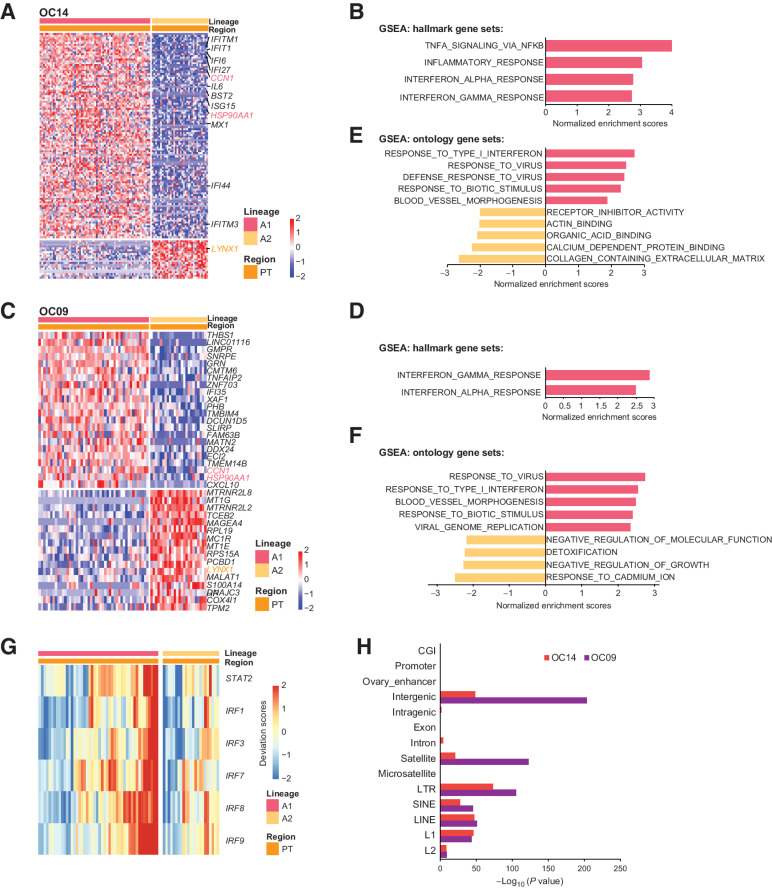
High-grade serous cancer (HGSC) is the most common subtype of ovarian cancer. HGSC is highly aggressive with poor patient outcomes, and a deeper understanding of HGSC tumorigenesis could help guide future treatment development. To systematically characterize the underlying pathologic mechanisms and intratumoral heterogeneity in human HGSC, we used an optimized single-cell multiomics sequencing technology to simultaneously analyze somatic copy-number alterations (SCNA), DNA methylation, chromatin accessibility, and transcriptome in individual cancer cells. Genes associated with interferon signaling, metallothioneins, and metabolism were commonly upregulated in ovarian cancer cells. Integrated multiomics analyses revealed that upregulation of interferon signaling and metallothioneins was influenced by both demethylation of their promoters and hypomethylation of satellites and LINE1, and potential key transcription factors regulating glycolysis using chromatin accessibility data were uncovered. In addition, gene expression and DNA methylation displayed similar patterns in matched primary and abdominal metastatic tumor cells of the same genetic lineage, suggesting that metastatic cells potentially preexist in the subclones of primary tumors. Finally, the lineages of cancer cells with higher residual DNA methylation levels and upregulated expression of CCN1 and HSP90AA1 presented greater metastatic potential. This study characterizes the critical genetic, epigenetic, and transcriptomic features and their mutual regulatory relationships in ovarian cancer, providing valuable resources for identifying new molecular mechanisms and potential therapeutic targets for HGSC.
Significance: Integrated analysis of multiomic changes and epigenetic regulation in high-grade serous ovarian cancer provides insights into the molecular characteristics of this disease, which could help improve diagnosis and treatment.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
Similar articles
-
[Significance and expression of PAX8, PAX2, p53 and RAS in ovary and fallopian tubes to origin of ovarian high grade serous carcinoma].Zhonghua Fu Chan Ke Za Zhi. 2017 Oct 25;52(10):687-696. doi: 10.3760/cma.j.issn.0529-567X.2017.10.008. Zhonghua Fu Chan Ke Za Zhi. 2017. PMID: 29060967 Chinese.
-
Epigenetic activation of POTE genes in ovarian cancer.Epigenetics. 2019 Feb;14(2):185-197. doi: 10.1080/15592294.2019.1581590. Epub 2019 Mar 4. Epigenetics. 2019. PMID: 30764732 Free PMC article.
-
Single-cell RNA-sequencing analysis reveals divergent transcriptome events between platinum-sensitive and platinum-resistant high-grade serous ovarian carcinoma.J Gene Med. 2023 Oct;25(10):e3504. doi: 10.1002/jgm.3504. Epub 2023 Apr 26. J Gene Med. 2023. PMID: 36994597
-
Peritoneal dissemination of high-grade serous ovarian cancer: pivotal roles of chromosomal instability and epigenetic dynamics.J Gynecol Oncol. 2022 Sep;33(5):e83. doi: 10.3802/jgo.2022.33.e83. J Gynecol Oncol. 2022. PMID: 36032027 Free PMC article. Review.
-
Multi-omics in high-grade serous ovarian cancer: Biomarkers from genome to the immunome.Am J Reprod Immunol. 2018 Aug;80(2):e12975. doi: 10.1111/aji.12975. Epub 2018 May 21. Am J Reprod Immunol. 2018. PMID: 29781548 Review.
Cited by
-
TIMM8B promotes oxidative phosphorylation and glycolysis by inhibiting the mtROS/ASK1/JNK signaling pathway in ovarian cancer.Biol Direct. 2025 Jul 1;20(1):75. doi: 10.1186/s13062-025-00663-6. Biol Direct. 2025. PMID: 40597377 Free PMC article.
-
Tumoroid model recreates clinically relevant phenotypes of high grade serous ovarian cancer (HGSC) cells, carcinoma associated fibroblasts, and macrophages.Res Sq [Preprint]. 2025 Jun 19:rs.3.rs-6614892. doi: 10.21203/rs.3.rs-6614892/v1. Res Sq. 2025. PMID: 40585272 Free PMC article. Preprint.
-
Single-cell transcriptomics in ovarian cancer identify a metastasis-associated cell cluster overexpressed RAB13.J Transl Med. 2023 Apr 12;21(1):254. doi: 10.1186/s12967-023-04094-7. J Transl Med. 2023. PMID: 37046345 Free PMC article.
-
Integrated multi-omics analysis reveals the immunotherapeutic significance of tumor cells with high FN1 expression in ovarian cancer.Front Mol Biosci. 2025 Jun 19;12:1611964. doi: 10.3389/fmolb.2025.1611964. eCollection 2025. Front Mol Biosci. 2025. PMID: 40612060 Free PMC article.
-
scRNA-seq Can Identify Different Cell Populations in Ovarian Cancer Bulk RNA-seq Experiments.Int J Mol Sci. 2025 Aug 4;26(15):7512. doi: 10.3390/ijms26157512. Int J Mol Sci. 2025. PMID: 40806640 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
