

De novo designed protein inhibitors of amyloid aggregation and seeding
- PMID: 35969734
- PMCID: PMC9407671
- DOI: 10.1073/pnas.2206240119
De novo designed protein inhibitors of amyloid aggregation and seeding
Abstract
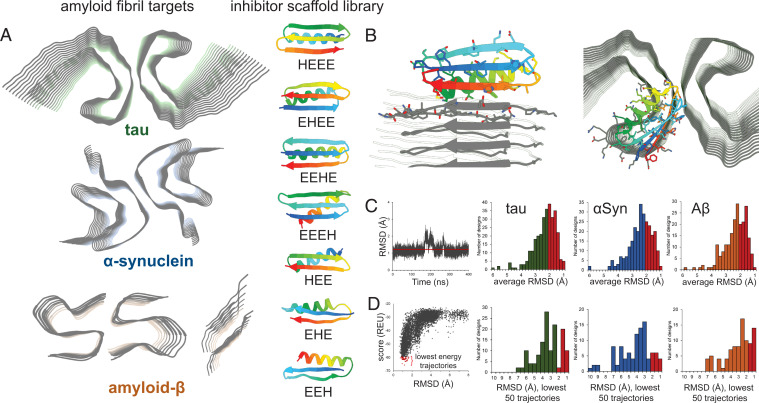
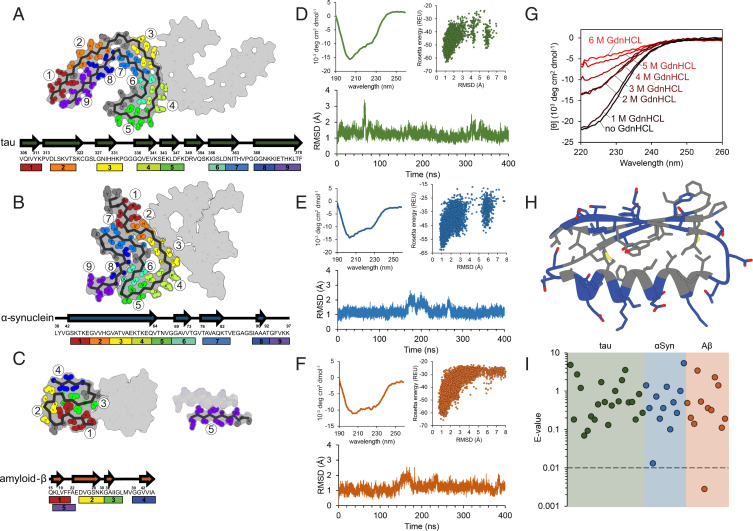
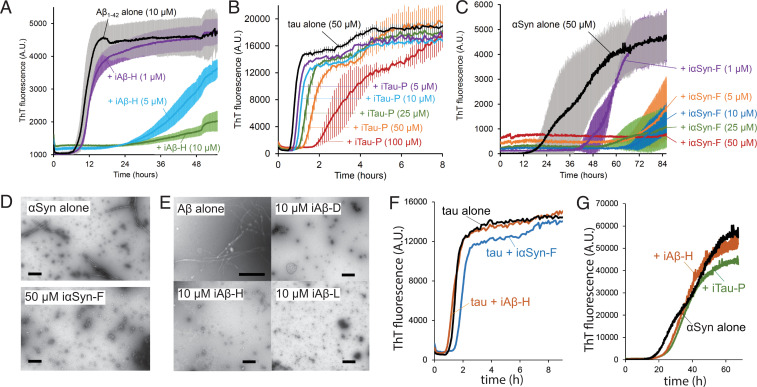
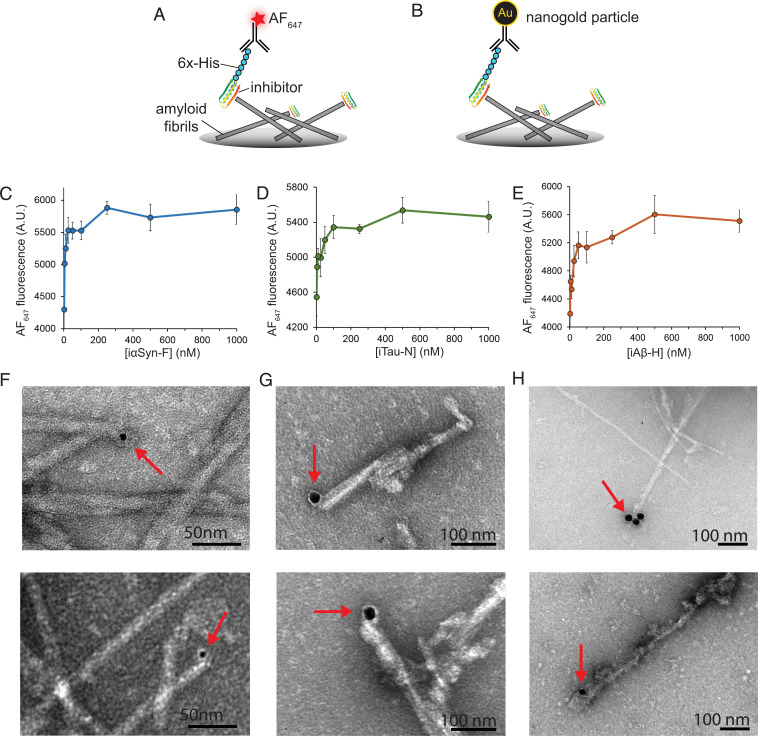
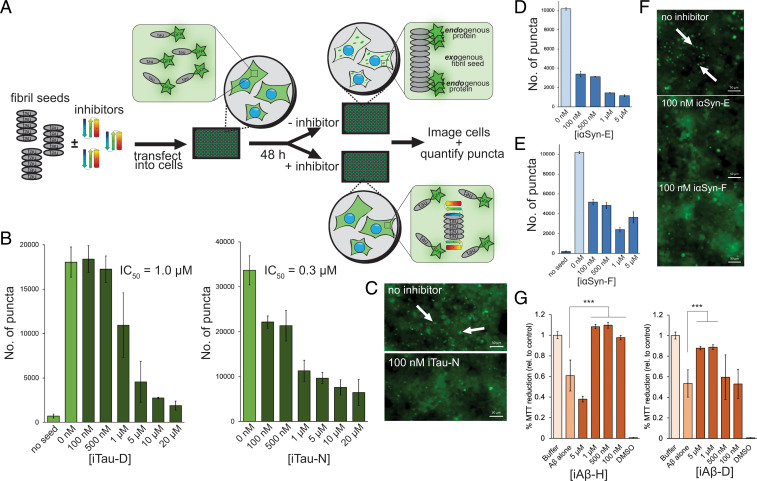
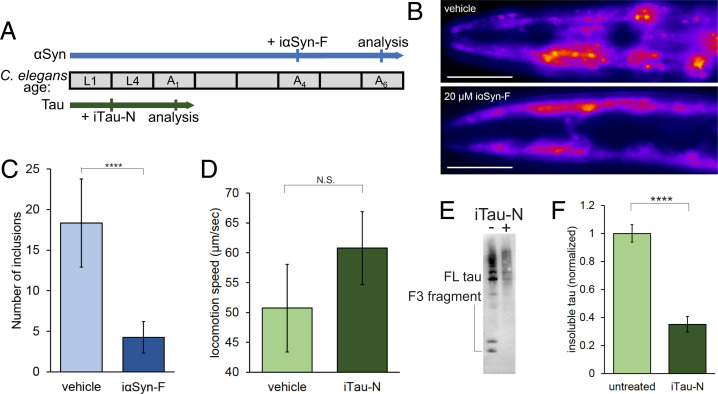
Neurodegenerative diseases are characterized by the pathologic accumulation of aggregated proteins. Known as amyloid, these fibrillar aggregates include proteins such as tau and amyloid-β (Aβ) in Alzheimer's disease (AD) and alpha-synuclein (αSyn) in Parkinson's disease (PD). The development and spread of amyloid fibrils within the brain correlates with disease onset and progression, and inhibiting amyloid formation is a possible route toward therapeutic development. Recent advances have enabled the determination of amyloid fibril structures to atomic-level resolution, improving the possibility of structure-based inhibitor design. In this work, we use these amyloid structures to design inhibitors that bind to the ends of fibrils, "capping" them so as to prevent further growth. Using de novo protein design, we develop a library of miniprotein inhibitors of 35 to 48 residues that target the amyloid structures of tau, Aβ, and αSyn. Biophysical characterization of top in silico designed inhibitors shows they form stable folds, have no sequence similarity to naturally occurring proteins, and specifically prevent the aggregation of their targeted amyloid-prone proteins in vitro. The inhibitors also prevent the seeded aggregation and toxicity of fibrils in cells. In vivo evaluation reveals their ability to reduce aggregation and rescue motor deficits in Caenorhabditis elegans models of PD and AD.
Keywords: alpha-synuclein; amyloid; amyloid-beta; protein design; tau.
Conflict of interest statement
Competing interest statement: D.S.E. is Science Advisory Board chair and equity holder of ADRx, Inc.
Figures
References
-
- Bloom G. S., Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508 (2014). - PubMed
-
- Wang Q., Yu X., Li L., Zheng J., Inhibition of amyloid-β aggregation in Alzheimer’s disease. Curr. Pharm. Des. 20, 1223–1243 (2014). - PubMed
-
- Bulic B., et al. , Development of tau aggregation inhibitors for Alzheimer’s disease. Angew. Chem. Int. Ed. Engl. 48, 1740–1752 (2009). - PubMed
