

Repertoire-scale measures of antigen binding
- PMID: 35969768
- PMCID: PMC9407674
- DOI: 10.1073/pnas.2203505119
Repertoire-scale measures of antigen binding
Abstract
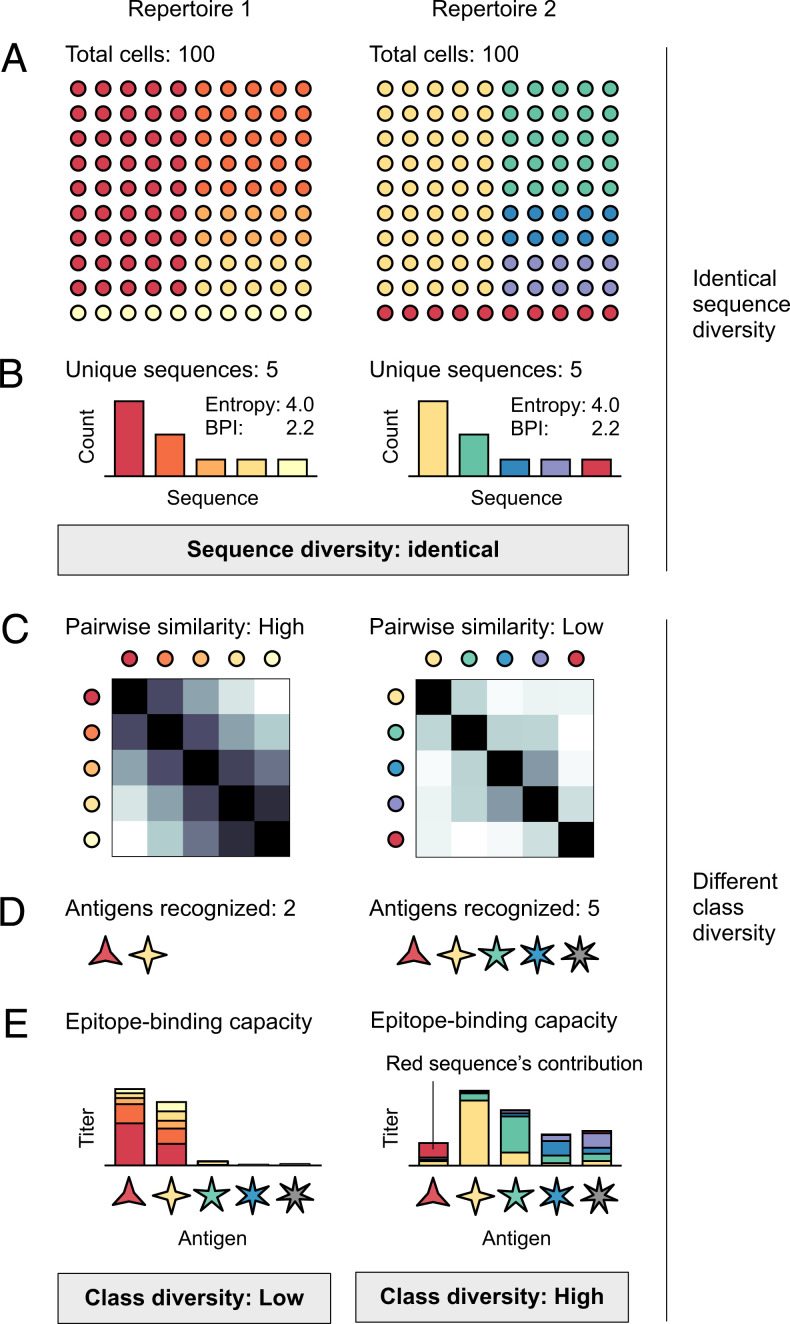
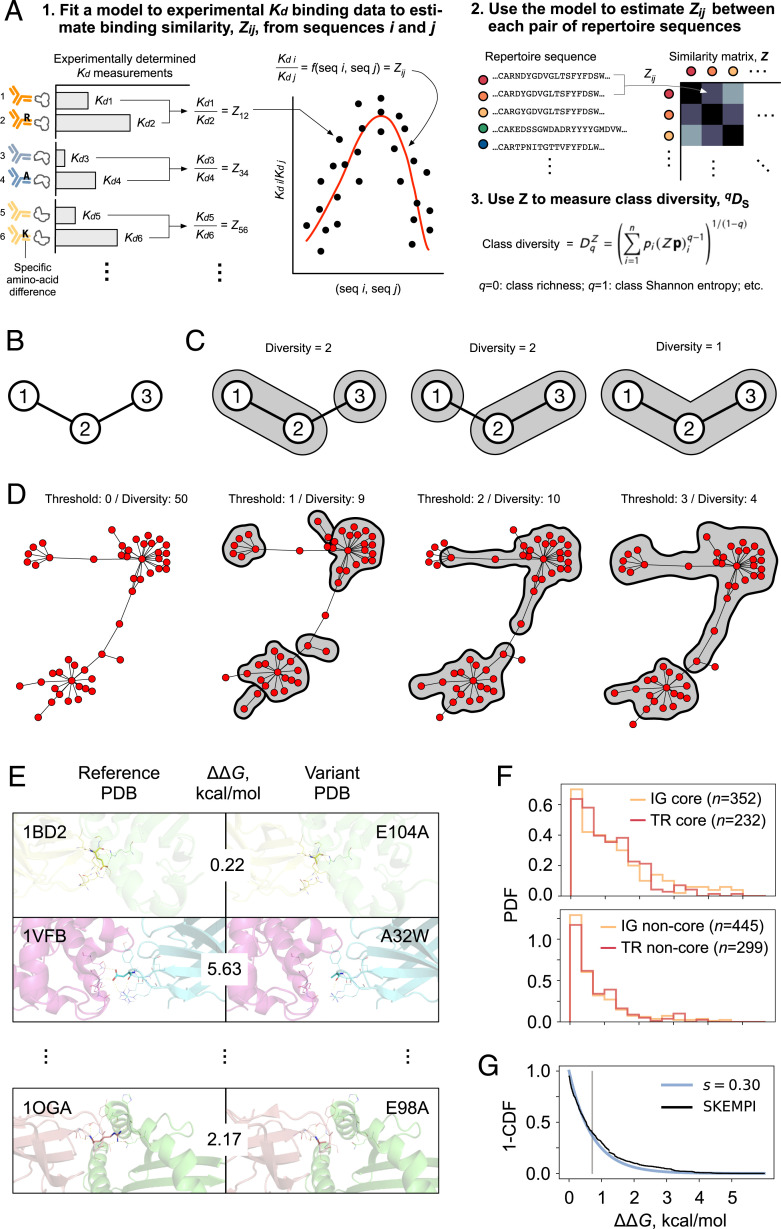
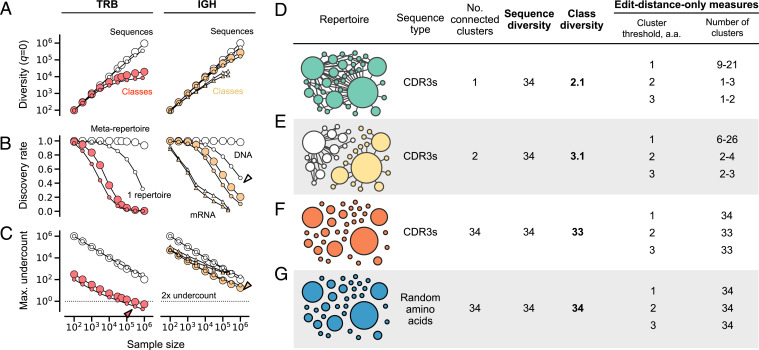
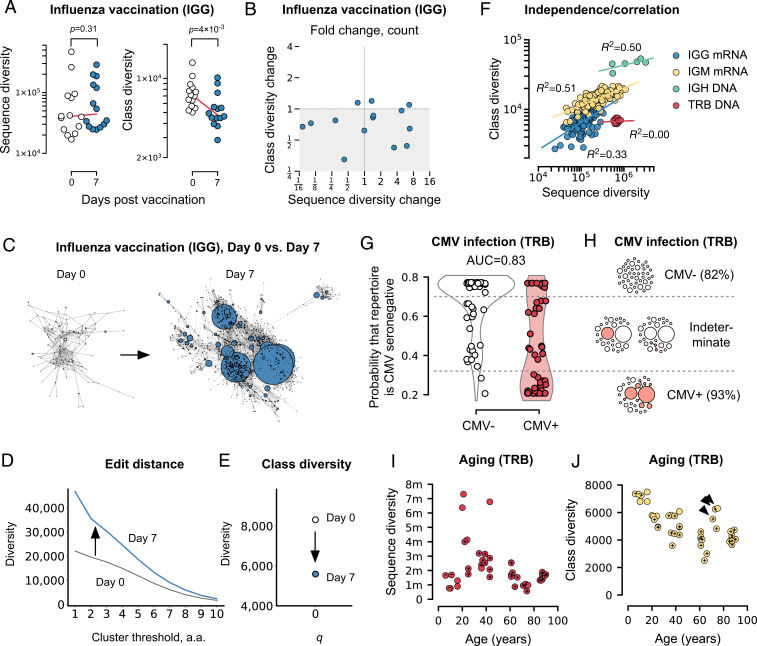
Antibodies and T cell receptors (TCRs) are the fundamental building blocks of adaptive immunity. Repertoire-scale functionality derives from their epitope-binding properties, just as macroscopic properties like temperature derive from microscopic molecular properties. However, most approaches to repertoire-scale measurement, including sequence diversity and entropy, are not based on antibody or TCR function in this way. Thus, they potentially overlook key features of immunological function. Here we present a framework that describes repertoires in terms of the epitope-binding properties of their constituent antibodies and TCRs, based on analysis of thousands of antibody-antigen and TCR-peptide-major-histocompatibility-complex binding interactions and over 400 high-throughput repertoires. We show that repertoires consist of loose overlapping classes of antibodies and TCRs with similar binding properties. We demonstrate the potential of this framework to distinguish specific responses vs. bystander activation in influenza vaccinees, stratify cytomegalovirus (CMV)-infected cohorts, and identify potential immunological "super-agers." Classes add a valuable dimension to the assessment of immune function.
Keywords: B cell repertoires; Gibbs free energy; T cell repertoires; antigen binding; immunological diversity.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Hill M. O., Diversity and evenness: A unifying notation and its consequences. Ecology 54, 427–432 (1973).
-
- Britanova O. V., et al. , Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J. Immunol. 192, 2689–2698 (2014). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
