

Deep learning predicts DNA methylation regulatory variants in the human brain and elucidates the genetics of psychiatric disorders
- PMID: 35969790
- PMCID: PMC9407663
- DOI: 10.1073/pnas.2206069119
Deep learning predicts DNA methylation regulatory variants in the human brain and elucidates the genetics of psychiatric disorders
Abstract
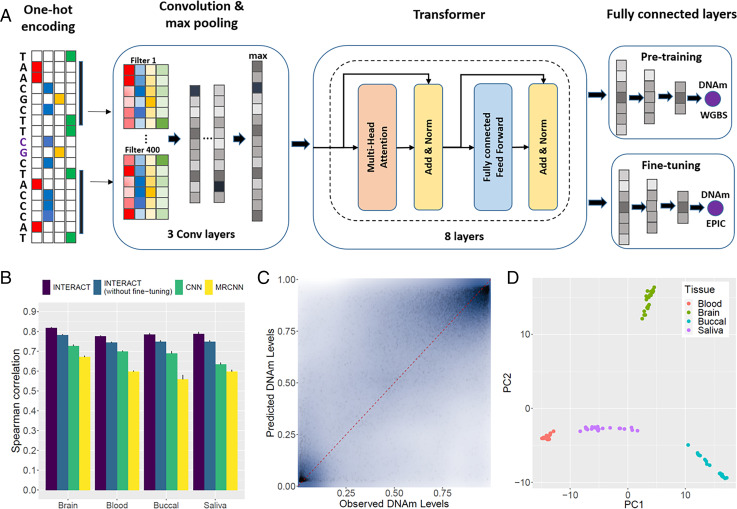
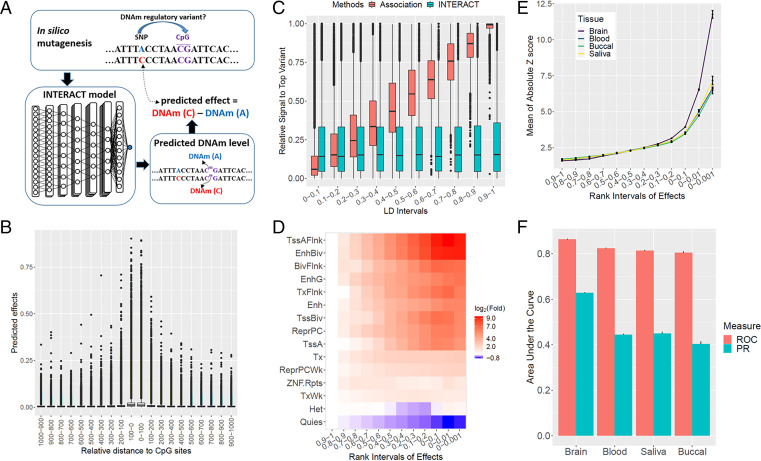
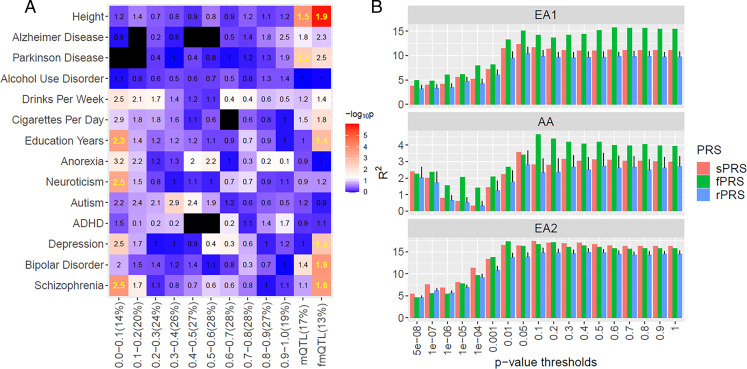
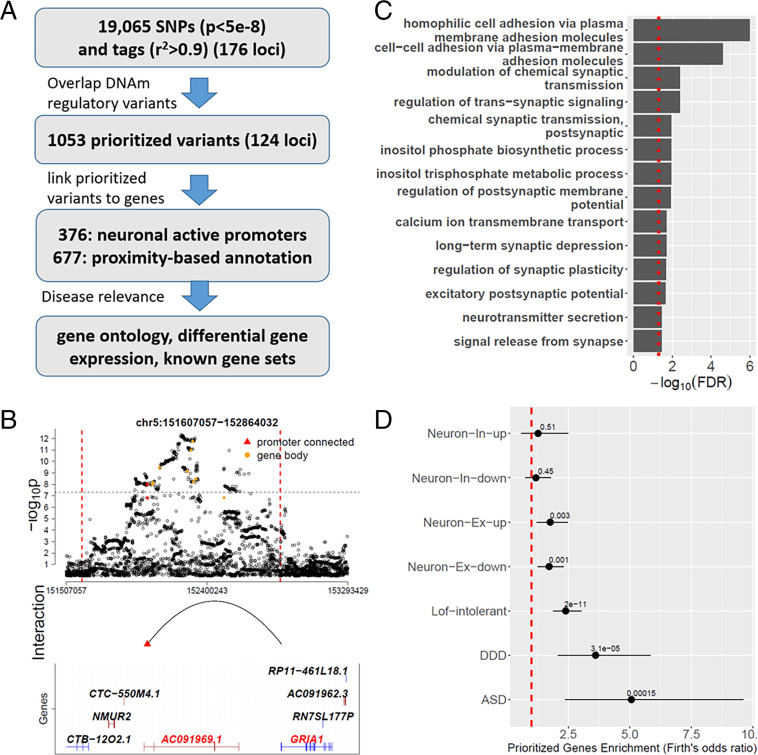
There is growing evidence for the role of DNA methylation (DNAm) quantitative trait loci (mQTLs) in the genetics of complex traits, including psychiatric disorders. However, due to extensive linkage disequilibrium (LD) of the genome, it is challenging to identify causal genetic variations that drive DNAm levels by population-based genetic association studies. This limits the utility of mQTLs for fine-mapping risk loci underlying psychiatric disorders identified by genome-wide association studies (GWAS). Here we present INTERACT, a deep learning model that integrates convolutional neural networks with transformer, to predict effects of genetic variations on DNAm levels at CpG sites in the human brain. We show that INTERACT-derived DNAm regulatory variants are not confounded by LD, are concentrated in regulatory genomic regions in the human brain, and are convergent with mQTL evidence from genetic association analysis. We further demonstrate that predicted DNAm regulatory variants are enriched for heritability of brain-related traits and improve polygenic risk prediction for schizophrenia across diverse ancestry samples. Finally, we applied predicted DNAm regulatory variants for fine-mapping schizophrenia GWAS risk loci to identify potential novel risk genes. Our study shows the power of a deep learning approach to identify functional regulatory variants that may elucidate the genetic basis of complex traits.
Keywords: DNA methylation quantitative trait loci (mQTL); GWAS; convolutional neural network (CNN); regulatory variants; transformer.
Conflict of interest statement
Competing interest statement: A.E.J. Is a current employee and shareholder of Neumora Therapeutics.
Figures
Similar articles
-
Deep learning predicts DNA methylation regulatory variants in specific brain cell types and enhances fine mapping for brain disorders.Sci Adv. 2025 Jan 3;11(1):eadn1870. doi: 10.1126/sciadv.adn1870. Epub 2025 Jan 1. Sci Adv. 2025. PMID: 39742481 Free PMC article.
-
Deep learning predicts DNA methylation regulatory variants in specific brain cell types and enhances fine mapping for brain disorders.bioRxiv [Preprint]. 2024 Jan 21:2024.01.18.576319. doi: 10.1101/2024.01.18.576319. bioRxiv. 2024. Update in: Sci Adv. 2025 Jan 3;11(1):eadn1870. doi: 10.1126/sciadv.adn1870. PMID: 38293210 Free PMC article. Updated. Preprint.
-
Genome-wide sequencing-based identification of methylation quantitative trait loci and their role in schizophrenia risk.Nat Commun. 2021 Sep 2;12(1):5251. doi: 10.1038/s41467-021-25517-3. Nat Commun. 2021. PMID: 34475392 Free PMC article.
-
Genetic-epigenetic interactions in cis: a major focus in the post-GWAS era.Genome Biol. 2017 Jun 19;18(1):120. doi: 10.1186/s13059-017-1250-y. Genome Biol. 2017. PMID: 28629478 Free PMC article. Review.
-
Genetic architecture of schizophrenia: a review of major advancements.Psychol Med. 2021 Oct;51(13):2168-2177. doi: 10.1017/S0033291720005334. Epub 2021 Feb 8. Psychol Med. 2021. PMID: 33550997 Review.
Cited by
-
Leveraging hierarchical structures for genetic block interaction studies using the hierarchical transformer.medRxiv [Preprint]. 2025 Feb 14:2024.11.18.24317486. doi: 10.1101/2024.11.18.24317486. medRxiv. 2025. PMID: 39606365 Free PMC article. Preprint.
-
MLACNN: an attention mechanism-based CNN architecture for predicting genome-wide DNA methylation.Theory Biosci. 2023 Nov;142(4):359-370. doi: 10.1007/s12064-023-00402-3. Epub 2023 Aug 30. Theory Biosci. 2023. PMID: 37648910 Free PMC article.
-
Deep learning imputes DNA methylation states in single cells and enhances the detection of epigenetic alterations in schizophrenia.Cell Genom. 2025 Mar 12;5(3):100774. doi: 10.1016/j.xgen.2025.100774. Epub 2025 Feb 21. Cell Genom. 2025. PMID: 39986279 Free PMC article.
-
scMeFormer: a transformer-based deep learning model for imputing DNA methylation states in single cells enhances the detection of epigenetic alterations in schizophrenia.bioRxiv [Preprint]. 2024 Jan 25:2024.01.25.577200. doi: 10.1101/2024.01.25.577200. bioRxiv. 2024. Update in: Cell Genom. 2025 Mar 12;5(3):100774. doi: 10.1016/j.xgen.2025.100774. PMID: 38328094 Free PMC article. Updated. Preprint.
-
Transformer Architecture and Attention Mechanisms in Genome Data Analysis: A Comprehensive Review.Biology (Basel). 2023 Jul 22;12(7):1033. doi: 10.3390/biology12071033. Biology (Basel). 2023. PMID: 37508462 Free PMC article. Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
