

ER stress and UPR in Alzheimer's disease: mechanisms, pathogenesis, treatments
- PMID: 35970828
- PMCID: PMC9378716
- DOI: 10.1038/s41419-022-05153-5
ER stress and UPR in Alzheimer's disease: mechanisms, pathogenesis, treatments
Abstract
Alzheimer's disease (AD) is a devastating neurodegenerative disorder characterized by gradual loss of memory and cognitive function, which constitutes a heavy burden on the healthcare system globally. Current therapeutics to interfere with the underlying disease process in AD is still under development. Although many efforts have centered on the toxic forms of Aβ to effectively tackle AD, considering the unsatisfactory results so far it is vital to examine other targets and therapeutic approaches as well. The endoplasmic reticulum (ER) stress refers to the build-up of unfolded or misfolded proteins within the ER, thus, perturbing the ER and cellular homeostasis. Emerging evidence indicates that ER stress contributes to the onset and development of AD. A thorough elucidation of ER stress machinery in AD pathology may help to open up new therapeutic avenues in the management of this devastating condition to relieve the cognitive dementia symptoms. Herein, we aim at deciphering the unique role of ER stress in AD pathogenesis, reviewing key findings, and existing controversy in an attempt to summarize plausible therapeutic interventions in the management of AD pathophysiology.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
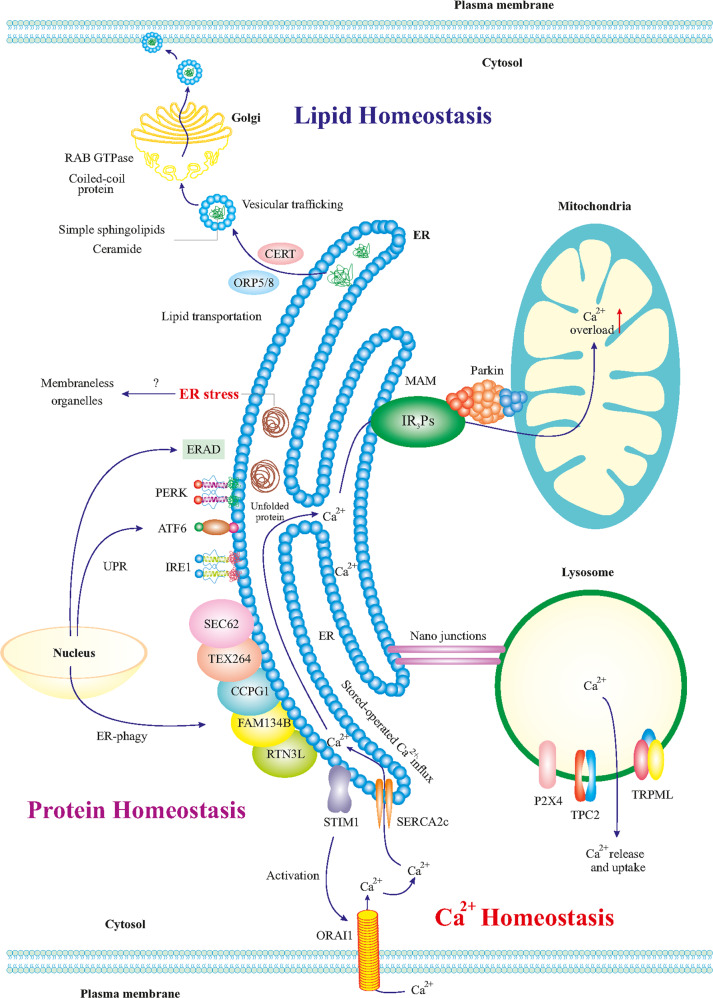
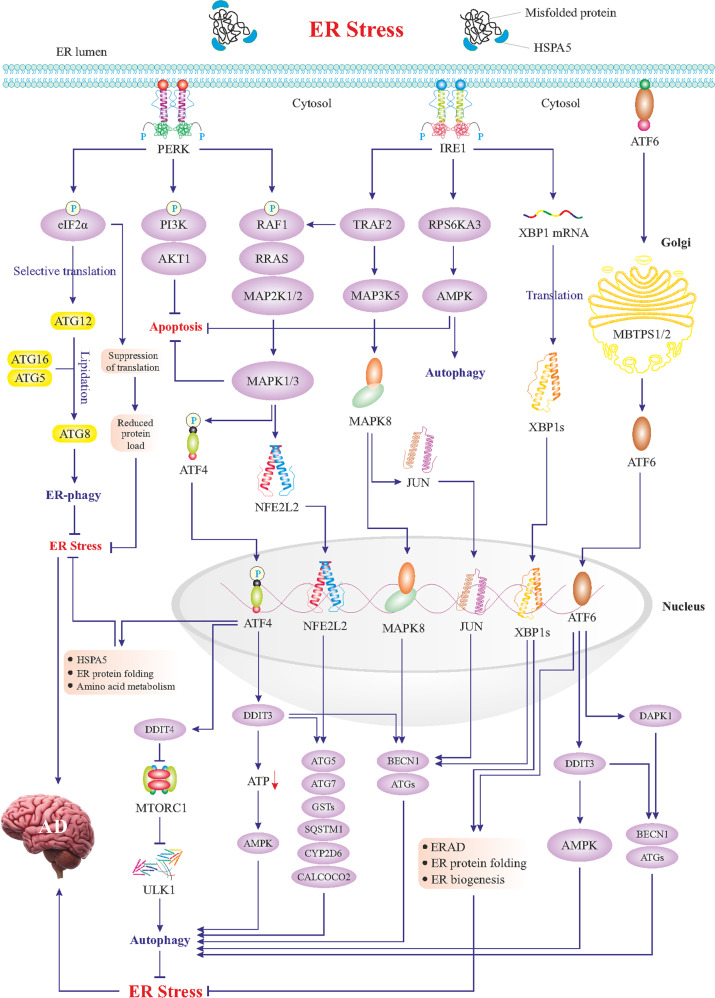
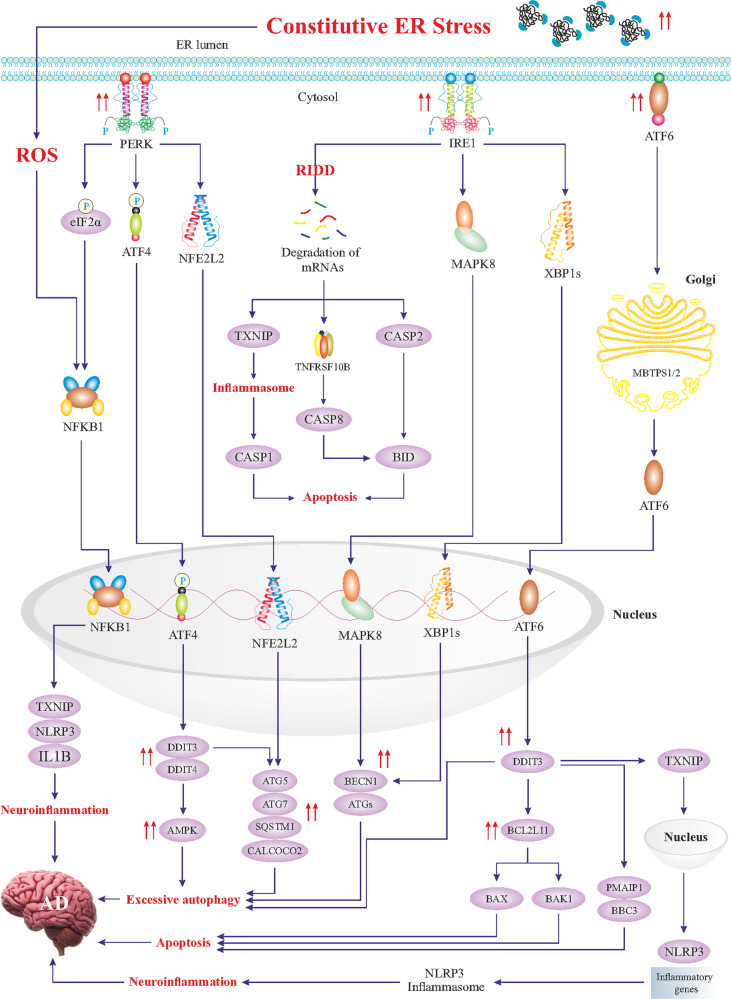
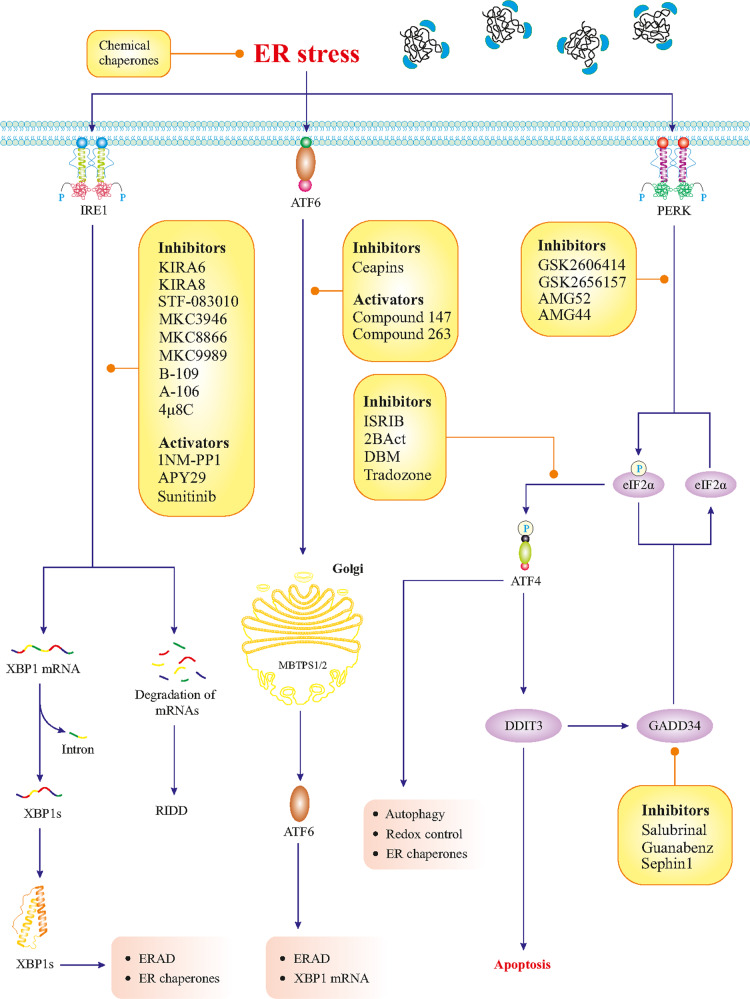
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
