

Understanding the mechanisms of HPV-related carcinogenesis: Implications for cell cycle dynamics
- PMID: 35973606
- PMCID: PMC9838640
- DOI: 10.1016/j.jtbi.2022.111235
Understanding the mechanisms of HPV-related carcinogenesis: Implications for cell cycle dynamics
Abstract
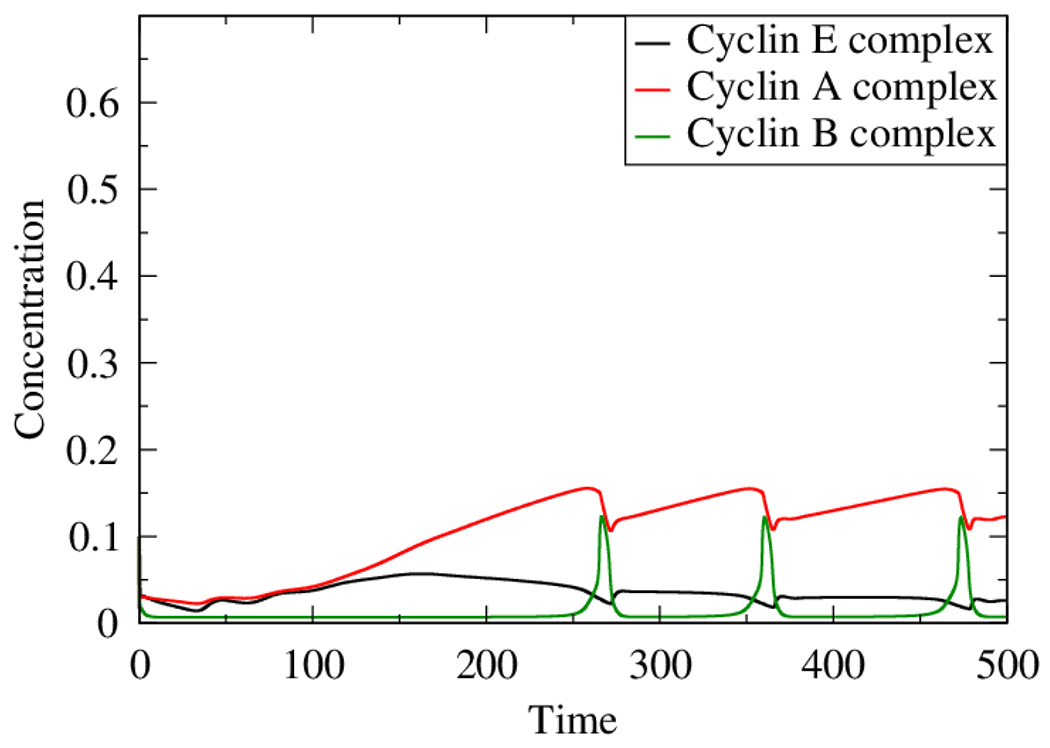
The role of human papillomavirus (HPV) as a causative agent for epithelial cancers is well-known, but many open questions remain regarding the downstream gene regulatory effects of viral proteins E6 and E7 on the cell cycle. Here, we extend a cell cycle model originally presented by Gérard and Goldbeter (2009) in order to capture the effects of E6 and E7 on key actors in the cell cycle. Results suggest that E6 is sufficient to reverse p53-induced quiescence, while E7 is sufficient to reverse p16INK4a-induced quiescence; both E6 and E7 are necessary when p53 and p16INK4a are both active. Moreover, E7 appears to play a role as a "growth factor substitute", inducing cell division in the absence of growth factor. Low levels of E7 may permit regular cell division, but the results suggest that higher levels of E7 dysregulate the cell cycle in ways that may destabilize the cellular genome. The mechanisms explored here provide opportunities for developing new treatment targets that take advantage of the cell cycle regulatory system to prevent HPV-related cancer effects.
Keywords: Cancer; Cell cycle; HPV; Mathematical modeling.
Copyright © 2022 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of Competing Interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures








Similar articles
-
TGF-beta-mediated cell cycle arrest of HPV16-immortalized human ectocervical cells correlates with decreased E6/E7 mRNA and increased p53 and p21(WAF-1) expression.Exp Cell Res. 2000 Aug 25;259(1):149-57. doi: 10.1006/excr.2000.4953. Exp Cell Res. 2000. PMID: 10942587
-
p16(INK4a) overexpression predicts translational active human papillomavirus infection in tonsillar cancer.Int J Cancer. 2010 Oct 1;127(7):1595-602. doi: 10.1002/ijc.25174. Int J Cancer. 2010. PMID: 20091864
-
Molecular mechanism of carcinogenesis by human papillomavirus-16.J Dermatol. 2000 Feb;27(2):73-86. doi: 10.1111/j.1346-8138.2000.tb02126.x. J Dermatol. 2000. PMID: 10721654 Review.
-
Immortalization of normal human embryonic fibroblasts by introduction of either the human papillomavirus type 16 E6 or E7 gene alone.Int J Cancer. 2003 Sep 1;106(3):301-9. doi: 10.1002/ijc.11219. Int J Cancer. 2003. PMID: 12845665
-
HPV DNA, E6/E7 mRNA, and p16INK4a detection in head and neck cancers: a systematic review and meta-analysis.Lancet Oncol. 2014 Nov;15(12):1319-31. doi: 10.1016/S1470-2045(14)70471-1. Epub 2014 Oct 16. Lancet Oncol. 2014. PMID: 25439690
Cited by
-
Development of human papillomavirus and its detection methods (Review).Exp Ther Med. 2024 Jul 31;28(4):382. doi: 10.3892/etm.2024.12671. eCollection 2024 Oct. Exp Ther Med. 2024. PMID: 39161614 Free PMC article. Review.
-
The Use of Intrinsic Disorder and Phosphorylation by Oncogenic Viral Proteins to Dysregulate the Host Cell Cycle Through Interaction with pRb.Viruses. 2025 Jun 10;17(6):835. doi: 10.3390/v17060835. Viruses. 2025. PMID: 40573426 Free PMC article. Review.
-
Survival Mechanisms of Metastatic Melanoma Cells: The Link between Glucocorticoids and the Nrf2-Dependent Antioxidant Defense System.Cells. 2023 Jan 26;12(3):418. doi: 10.3390/cells12030418. Cells. 2023. PMID: 36766760 Free PMC article. Review.
-
Human papillomavirus associated cervical lesion: pathogenesis and therapeutic interventions.MedComm (2020). 2023 Sep 14;4(5):e368. doi: 10.1002/mco2.368. eCollection 2023 Oct. MedComm (2020). 2023. PMID: 37719443 Free PMC article. Review.
References
-
- Jemal A, Simard EP, Dorell C, Noone A-M, Markowitz LE, Kohler B, Eheman C, Saraiya M, Bandi P, Saslow D, et al., “Annual report to the nation on the status of cancer, 1975–2009, featuring the burden and trends in human papillomavirus (hpv)–associated cancers and hpv vaccination coverage levels,” JNCI: Journal of the National Cancer Institute, vol. 105, no. 3, pp. 175–201, 2013. - PMC - PubMed
-
- C. for Disease Control, P. (CDC, et al. “Human papillomavirus-associated cancers-united states, 2004–2008.,” MMWR. Morbidity and mortality weekly report, vol. 61, p. 258, 2012. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
