

Role of necroptosis in airflow limitation in chronic obstructive pulmonary disease: focus on small-airway disease and emphysema
- PMID: 35973987
- PMCID: PMC9381515
- DOI: 10.1038/s41420-022-01154-7
Role of necroptosis in airflow limitation in chronic obstructive pulmonary disease: focus on small-airway disease and emphysema
Abstract
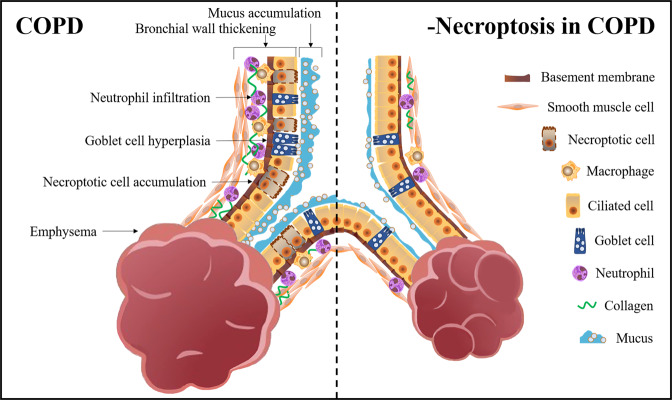
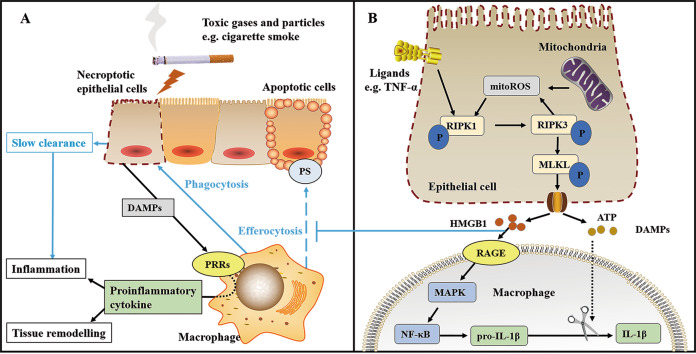
Airflow limitation with intractable progressive mechanisms is the main disease feature of chronic obstructive pulmonary disease (COPD). The pathological process of airflow limitation in COPD involves necroptosis, a form of programmed necrotic cell death with pro-inflammatory properties. In this paper, the correlations of small-airway disease and emphysema with airflow limitation in COPD were firstly reviewed; then, based on this, the effects of necroptosis on small-airway disease and emphysema were analysed, and the possible mechanisms of necroptosis causing airflow limitation in COPD were explored. The results showed that airflow limitation is caused by a combination of small-airway disease and emphysema. In addition, toxic particulate matter stimulates epithelial cells to trigger necroptosis, and necroptosis promotes the expulsion of cell contents, the abnormal hyperplasia of pro-inflammatory mediators and the insufficient clearance of dead cells by macrophages; these processes, coupled with the interaction of necroptosis and oxidative stress, collectively result in small-airway disease and emphysema in COPD.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- WHO. The top 10 causes of death. https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (2020).
-
- Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. https://goldcopd.org/wp-content/uploads/2021/11/GOLD-REPORT-2022-v1.1-22... (2022).
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
