

DDX39B drives colorectal cancer progression by promoting the stability and nuclear translocation of PKM2
- PMID: 35973989
- PMCID: PMC9381590
- DOI: 10.1038/s41392-022-01096-7
DDX39B drives colorectal cancer progression by promoting the stability and nuclear translocation of PKM2
Abstract
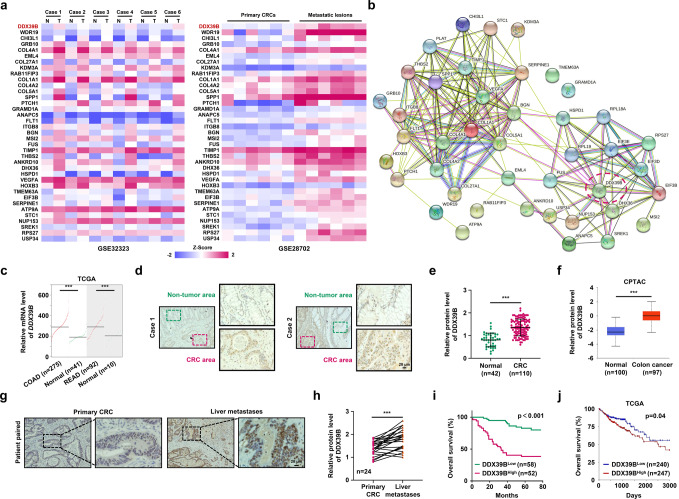
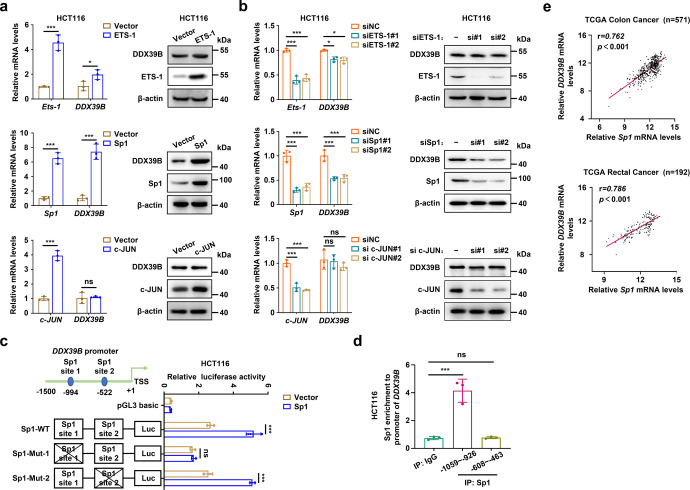
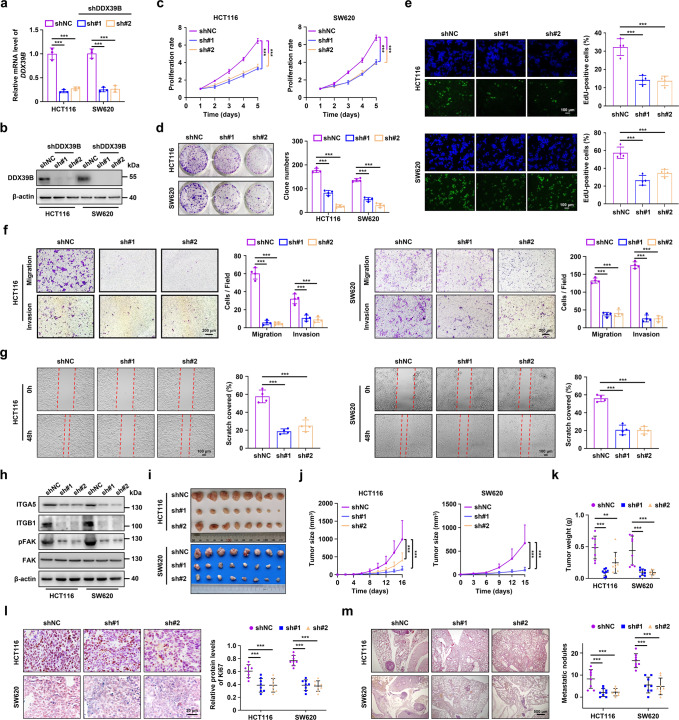
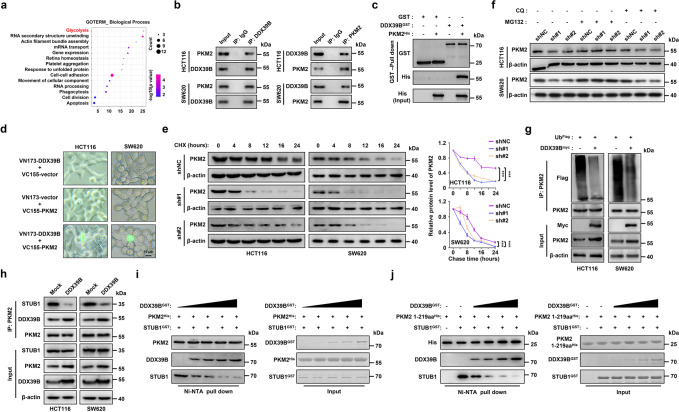
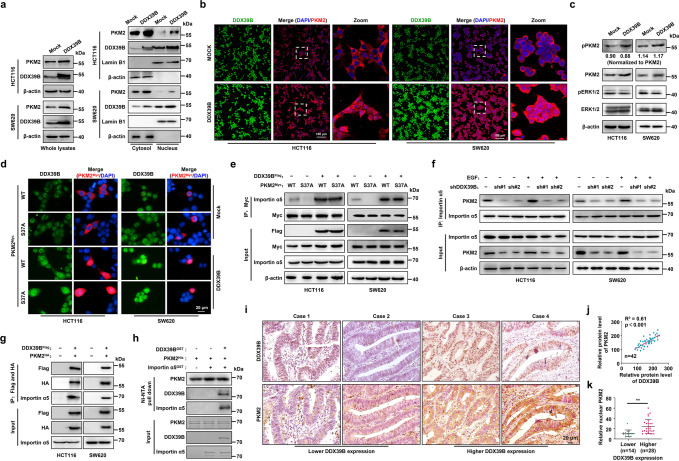
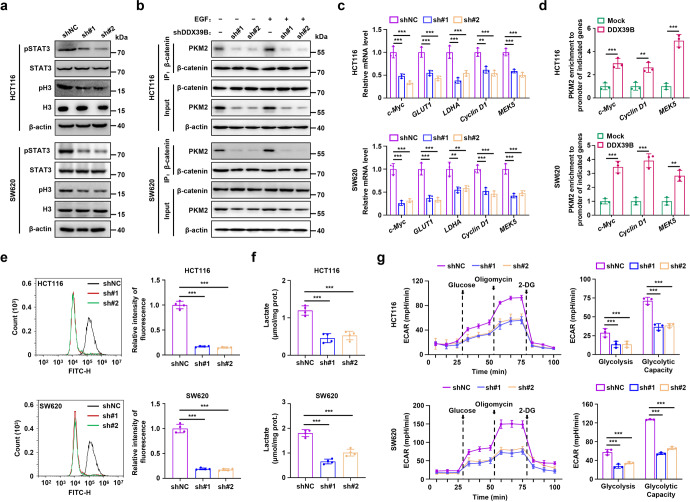
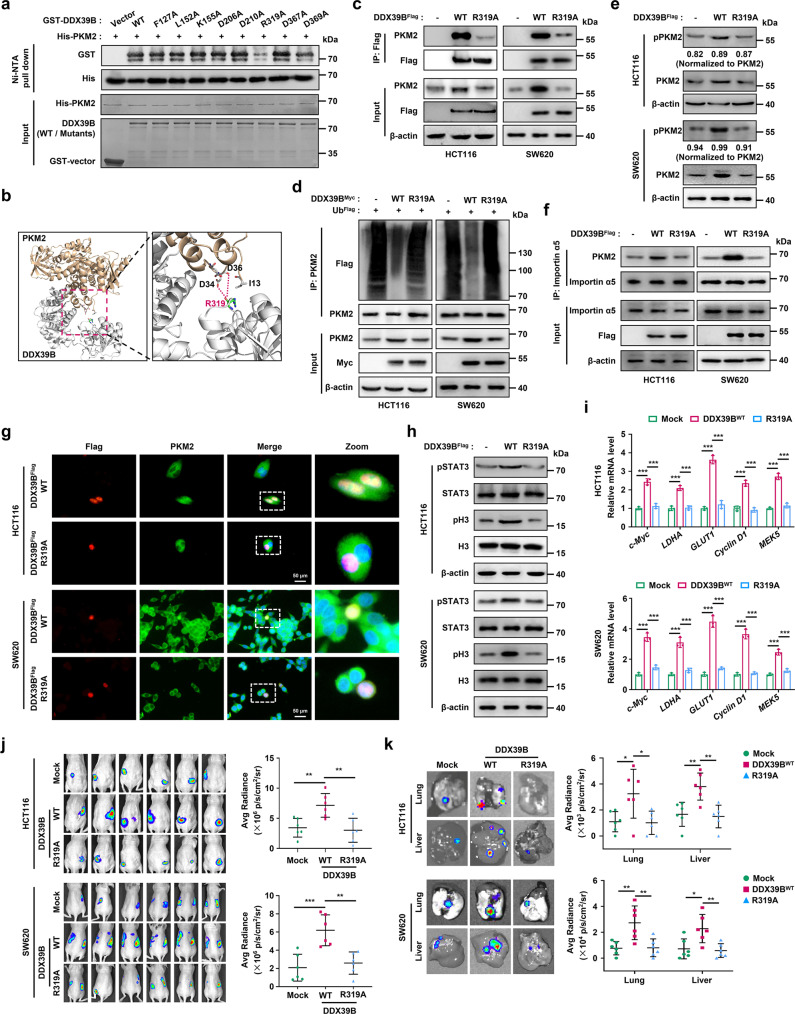
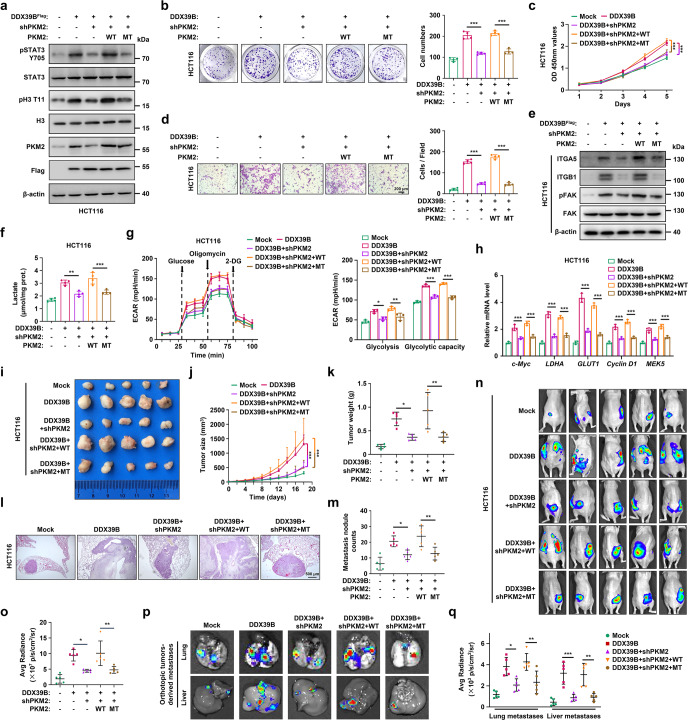
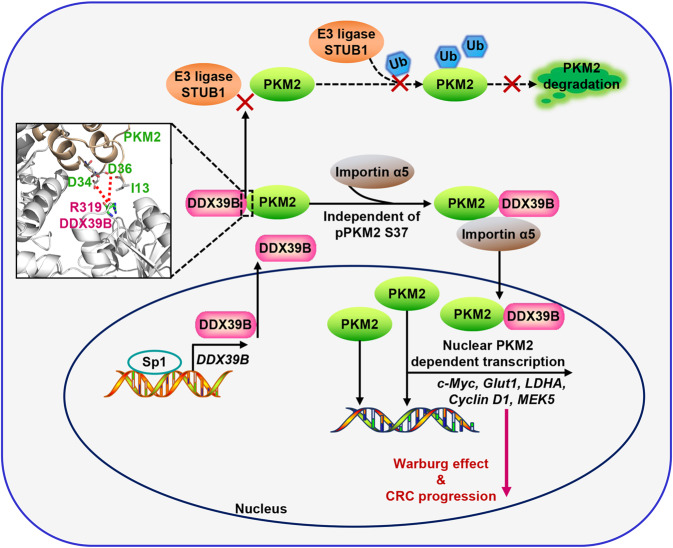
Metastasis is a major cause of colorectal cancer (CRC) mortality, but its molecular mechanisms are still not fully understood. Here, we show that upregulated DDX39B correlates with liver metastases and aggressive phenotypes in CRC. DDX39B is an independent prognostic factor associated with poor clinical outcome in CRC patients. We demonstrate that Sp1 potently activates DDX39B transcription by directly binding to the GC box of the DDX39B promoter in CRC cells. DDX39B overexpression augments the proliferation, migration, and invasion of CRC cells, while the opposite results are obtained in DDX39B-deficient CRC cells. Mechanistically, DDX39B interacts directly with and stabilizes PKM2 by competitively suppressing STUB1-mediated PKM2 ubiquitination and degradation. Importantly, DDX39B recruits importin α5 to accelerate the nuclear translocation of PKM2 independent of ERK1/2-mediated phosphorylation of PKM2, leading to the transactivation of oncogenes and glycolysis-related genes. Consequently, DDX39B enhances glucose uptake and lactate production to activate Warburg effect in CRC. We identify that Arg319 of DDX39B is required for PKM2 binding as well as PKM2 nuclear accumulation and for DDX39B to promote CRC growth and metastasis. In addition, blocking PKM2 nuclear translocation or treatment with glycolytic inhibitor 2-deoxy-D-glucose efficiently abolishes DDX39B-triggered malignant development in CRC. Taken together, our findings uncover a key role for DDX39B in modulating glycolytic reprogramming and aggressive progression, and implicate DDX39B as a potential therapeutic target in CRC.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
