

Macrophages as tools and targets in cancer therapy
- PMID: 35974096
- PMCID: PMC9380983
- DOI: 10.1038/s41573-022-00520-5
Macrophages as tools and targets in cancer therapy
Abstract
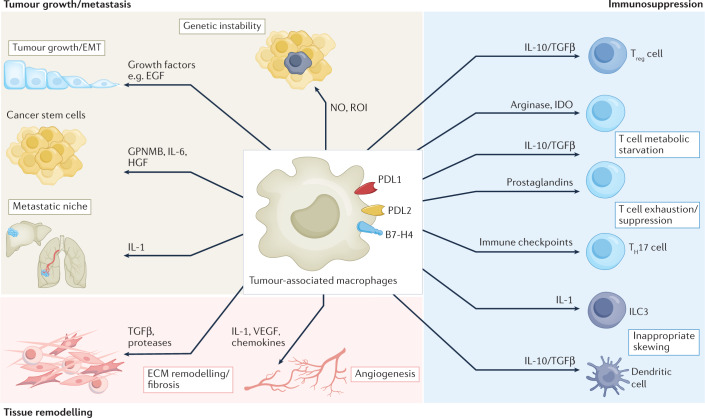
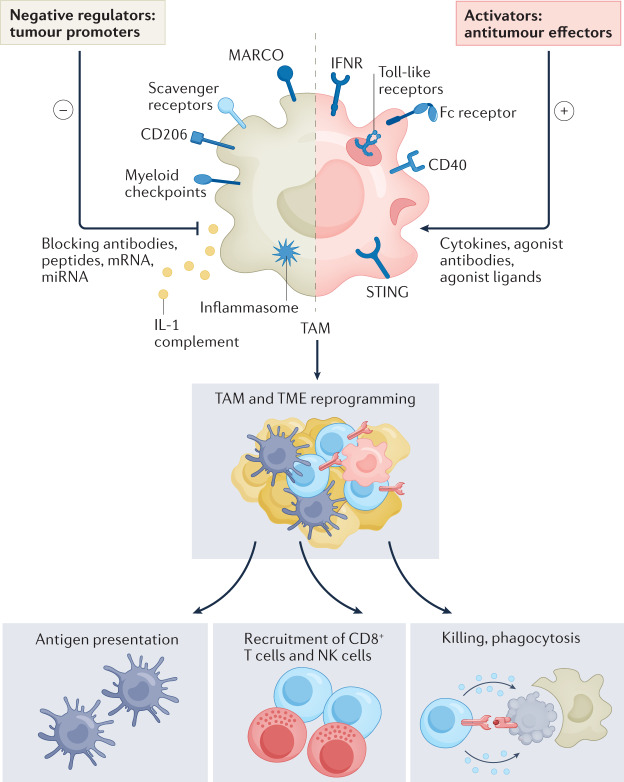
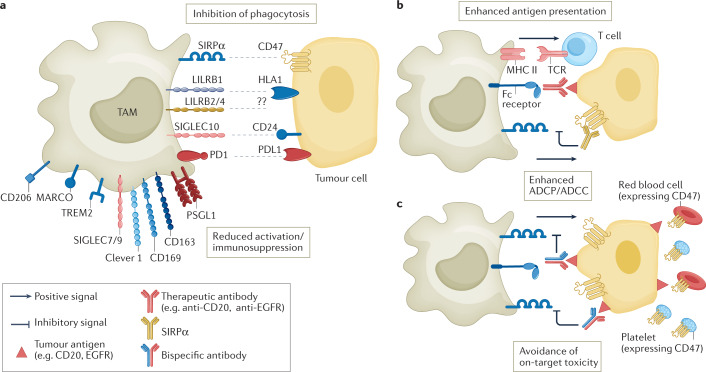
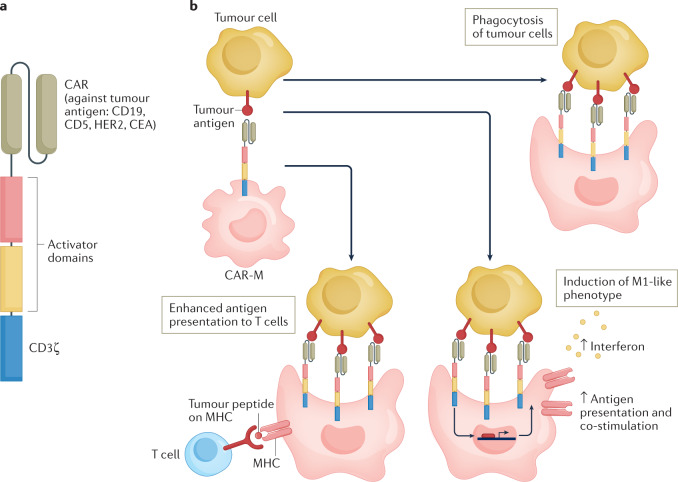
Tumour-associated macrophages are an essential component of the tumour microenvironment and have a role in the orchestration of angiogenesis, extracellular matrix remodelling, cancer cell proliferation, metastasis and immunosuppression, as well as in resistance to chemotherapeutic agents and checkpoint blockade immunotherapy. Conversely, when appropriately activated, macrophages can mediate phagocytosis of cancer cells and cytotoxic tumour killing, and engage in effective bidirectional interactions with components of the innate and adaptive immune system. Therefore, they have emerged as therapeutic targets in cancer therapy. Macrophage-targeting strategies include inhibitors of cytokines and chemokines involved in the recruitment and polarization of tumour-promoting myeloid cells as well as activators of their antitumorigenic and immunostimulating functions. Early clinical trials suggest that targeting negative regulators (checkpoints) of myeloid cell function indeed has antitumor potential. Finally, given the continuous recruitment of myelomonocytic cells into tumour tissues, macrophages are candidates for cell therapy with the development of chimeric antigen receptor effector cells. Macrophage-centred therapeutic strategies have the potential to complement, and synergize with, currently available tools in the oncology armamentarium.
© 2022. Springer Nature Limited.
Conflict of interest statement
A.M. has been a recipient of commercial research grants from Sigma Tau, Roche, Novartis, Compugen and Efranat, and has been a consultant/advisory board member/lecturer for Novartis, Roche, Ventana, Pierre Fabre, Verily, Abbvie, BMS, J&J, Compugen, Imcheck, Macrophage Therapeutics, AstraZeneca, Biovelocita, BG Fund, Third Rock, Verseau Therapeutics and Olatec Therapeutics. C.G. and P.A. are recipients of research grants from Imcheck and Macrophage Therapeutics. A.M. and C.G. are inventors of patents related to PTX3 and other innate immunity molecules. A.M., C.G. and P.A. receive royalties for reagents related to innate immunity.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
