

A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues
- PMID: 35974106
- PMCID: PMC9381717
- DOI: 10.1038/s41467-022-32552-1
A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues
Abstract
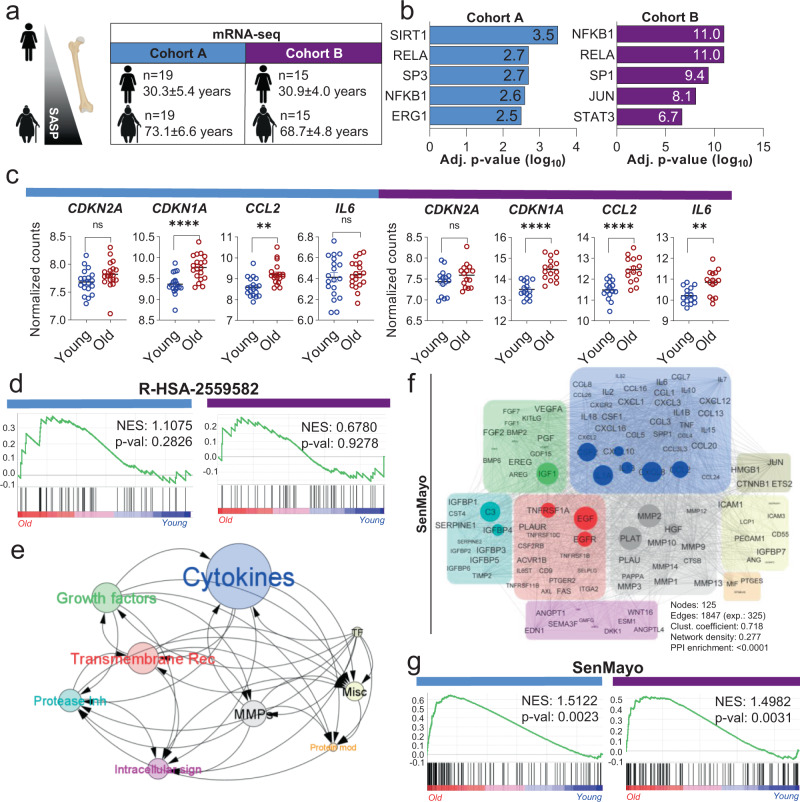
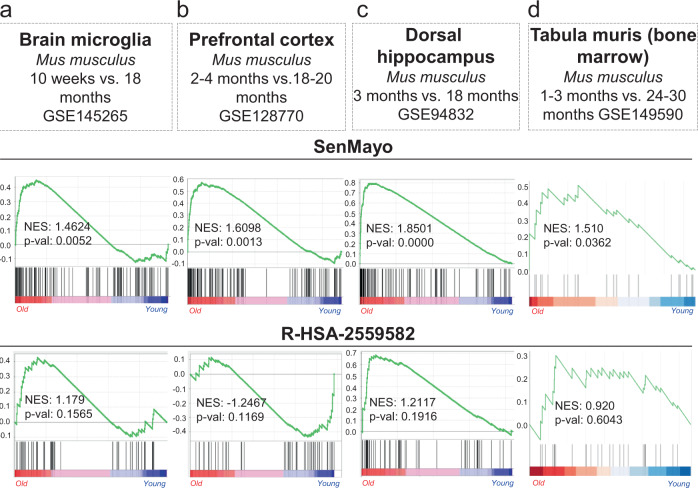
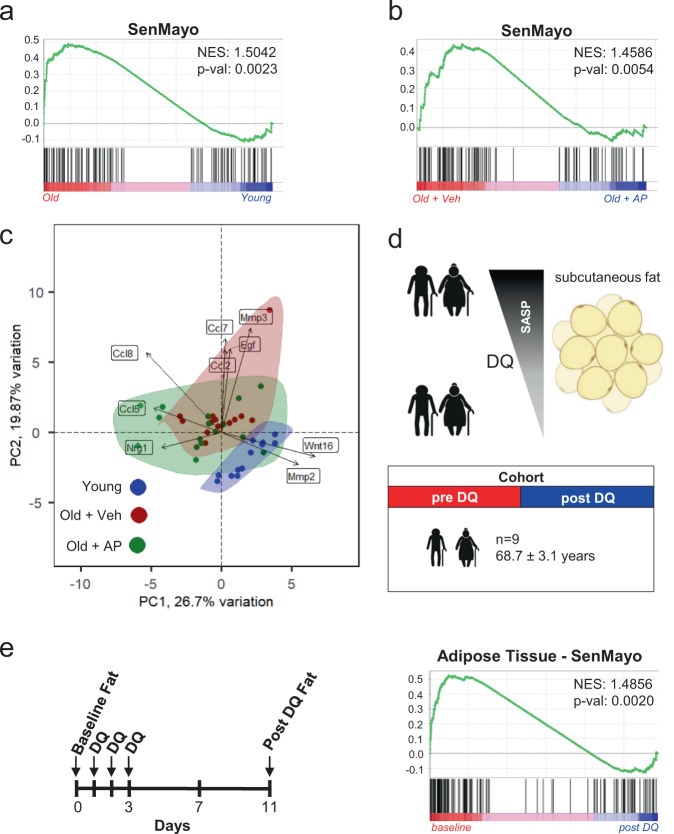
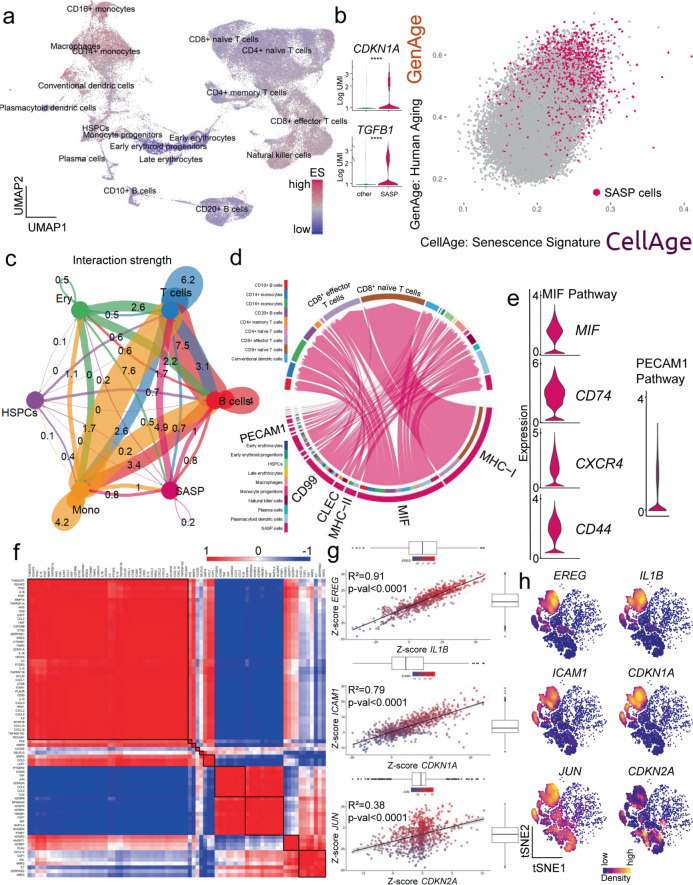
Although cellular senescence drives multiple age-related co-morbidities through the senescence-associated secretory phenotype, in vivo senescent cell identification remains challenging. Here, we generate a gene set (SenMayo) and validate its enrichment in bone biopsies from two aged human cohorts. We further demonstrate reductions in SenMayo in bone following genetic clearance of senescent cells in mice and in adipose tissue from humans following pharmacological senescent cell clearance. We next use SenMayo to identify senescent hematopoietic or mesenchymal cells at the single cell level from human and murine bone marrow/bone scRNA-seq data. Thus, SenMayo identifies senescent cells across tissues and species with high fidelity. Using this senescence panel, we are able to characterize senescent cells at the single cell level and identify key intercellular signaling pathways. SenMayo also represents a potentially clinically applicable panel for monitoring senescent cell burden with aging and other conditions as well as in studies of senolytic drugs.
© 2022. The Author(s).
Conflict of interest statement
J.L.K., T.T., and N.K.L. have a financial interest related to this research. Patents on senolytic drugs and their uses and SASP biomarkers are held by Mayo Clinic and the University of Minnesota. This research has been reviewed by the Mayo Clinic Conflict of Interest Review Board and was conducted in compliance with Mayo Clinic Conflict of Interest policies. The remaining authors declare no competing interests
Figures
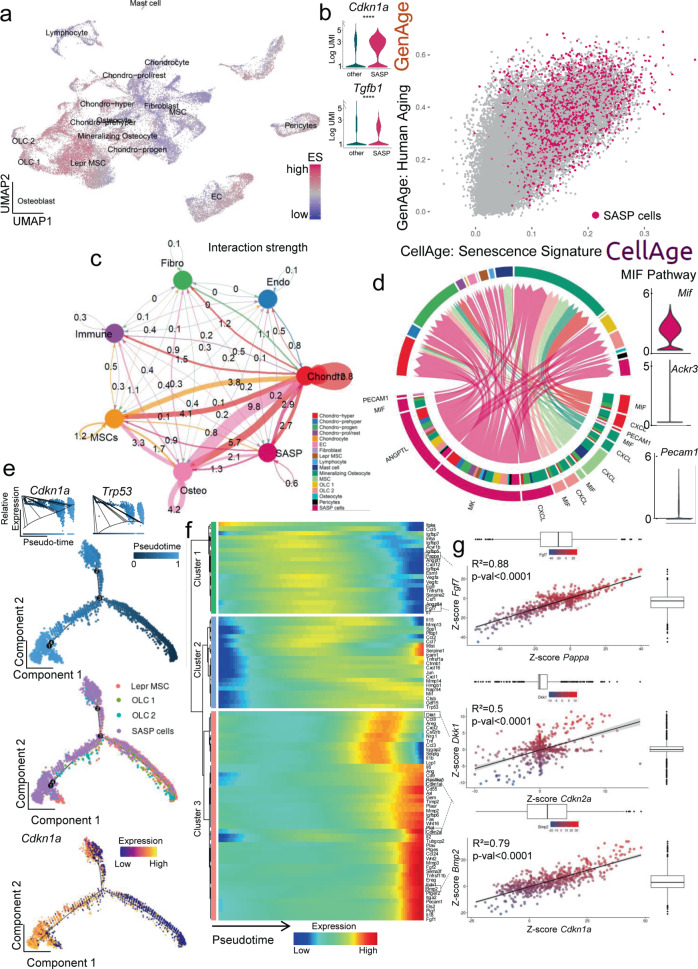
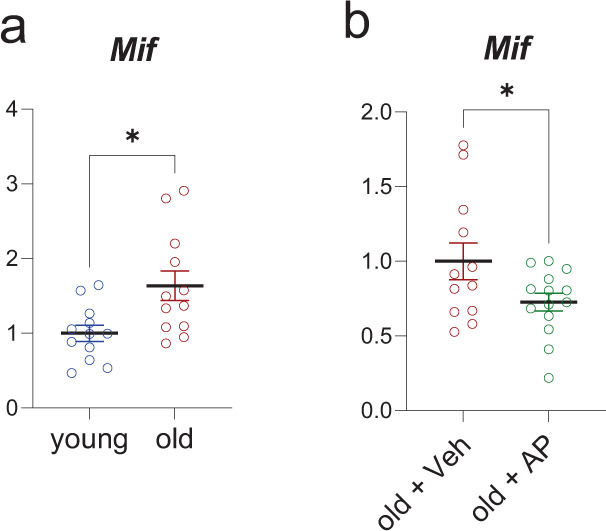
References
Publication types
MeSH terms
Grants and funding
- R01 DK128552/DK/NIDDK NIH HHS/United States
- T32 AG049672/AG/NIA NIH HHS/United States
- R37 AG013925/AG/NIA NIH HHS/United States
- R01 AG076515/AG/NIA NIH HHS/United States
- P30 AG013319/AG/NIA NIH HHS/United States
- R01 AG055529/AG/NIA NIH HHS/United States
- R21 AG065868/AG/NIA NIH HHS/United States
- R01 AG068048/AG/NIA NIH HHS/United States
- R56 AG060907/AG/NIA NIH HHS/United States
- K01 AR070241/AR/NIAMS NIH HHS/United States
- UG3 CA268103/CA/NCI NIH HHS/United States
- R33 AG061456/AG/NIA NIH HHS/United States
- R01 AG063707/AG/NIA NIH HHS/United States
- UH3 CA268103/CA/NCI NIH HHS/United States
- P01 AG062413/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
