

Aberrant Cholesterol Metabolism and Wnt/β-Catenin Signaling Coalesce via Frizzled5 in Supporting Cancer Growth
- PMID: 35975457
- PMCID: PMC9534957
- DOI: 10.1002/advs.202200750
Aberrant Cholesterol Metabolism and Wnt/β-Catenin Signaling Coalesce via Frizzled5 in Supporting Cancer Growth
Abstract
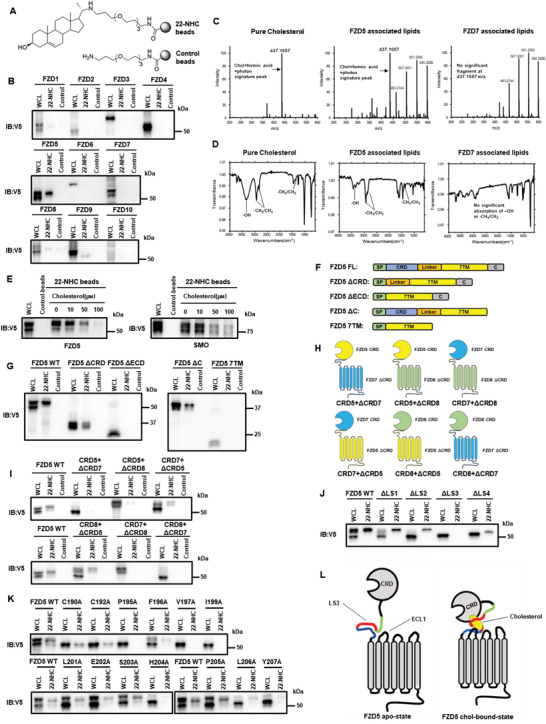
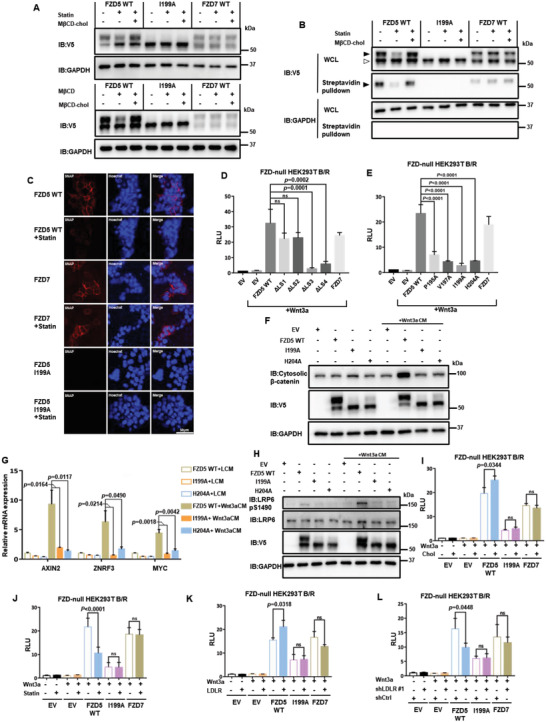
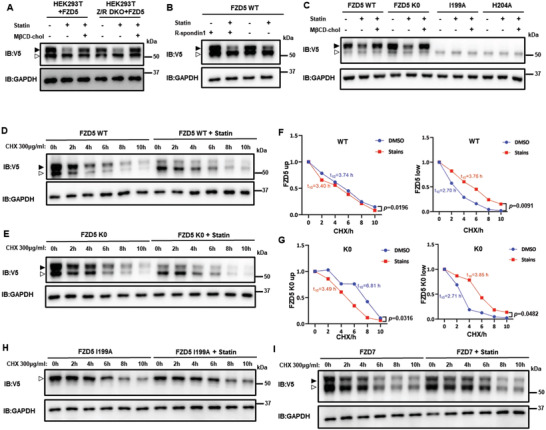
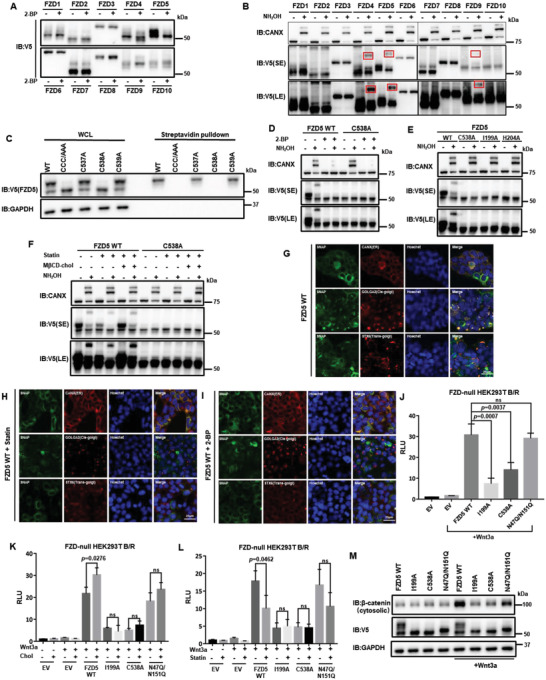
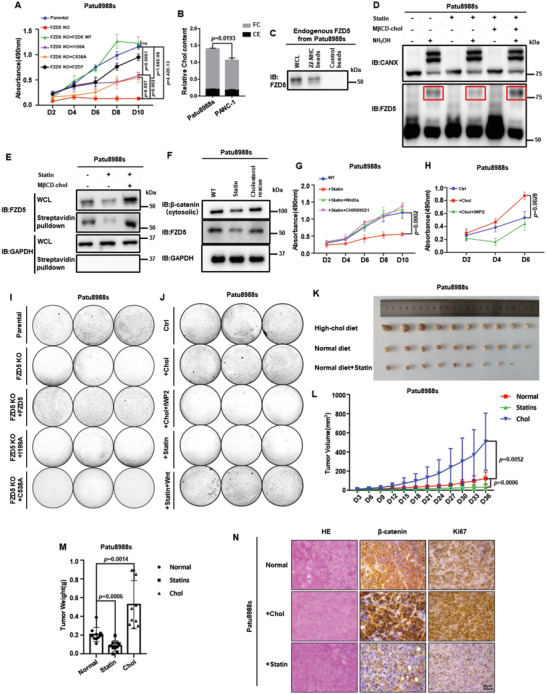
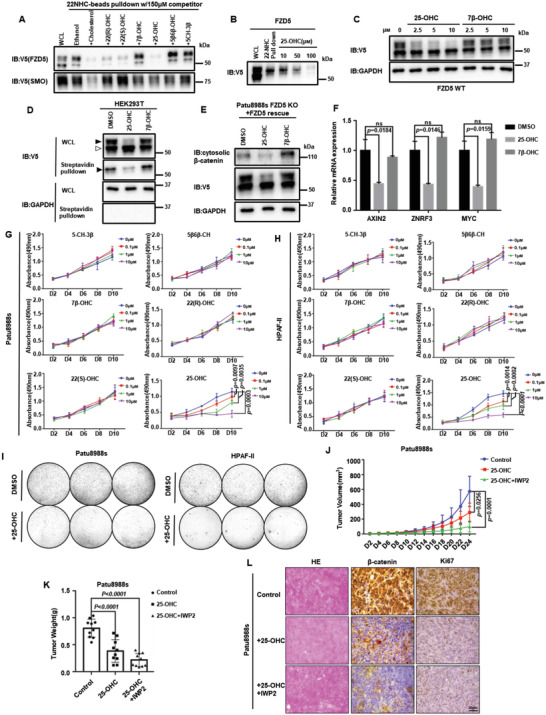
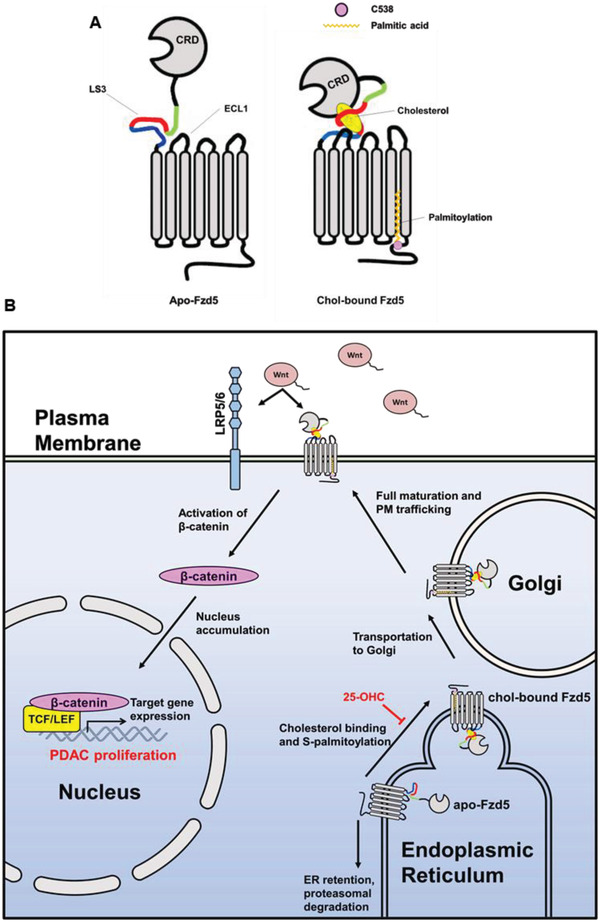
Frizzled (Fzd) proteins are Wnt receptors and play essential roles in development, homeostasis, and oncogenesis. How Wnt/Fzd signaling is coupled to physiological regulation remains unknown. Cholesterol is reported as a signaling molecule regulating morphogen such as Hedgehog signaling. Despite the elusiveness of the in-depth mechanism, it is well-established that pancreatic cancer specially requires abnormal cholesterol metabolism levels for growth. In this study, it is unexpectedly found that among ten Fzds, Fzd5 has a unique capacity to bind cholesterol specifically through its conserved extracellular linker region. Cholesterol-binding enables Fzd5 palmitoylation, which is indispensable for receptor maturation and trafficking to the plasma membrane. In Wnt-addicted pancreatic ductal adenocarcinoma (PDAC), cholesterol stimulates tumor growth via Fzd5-mediated Wnt/β-catenin signaling. A natural oxysterol, 25-hydroxylsterol competes with cholesterol and inhibits Fzd5 maturation and Wnt signaling, thereby alleviating PDAC growth. This cholesterol-receptor interaction and ensuing receptor lipidation uncover a novel mechanism by which Fzd5 acts as a cholesterol sensor and pivotal connection coupling lipid metabolism to morphogen signaling. These findings further suggest that cholesterol-targeting may provide new therapeutic opportunities for treating Wnt-dependent cancers.
Keywords: Wnt/β-catenin signaling; Frizzled receptor; cholesterol; pancreatic cancer.
© 2022 The Authors. Advanced Science published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Genome-wide CRISPR screens reveal a Wnt-FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors.Nat Med. 2017 Jan;23(1):60-68. doi: 10.1038/nm.4219. Epub 2016 Nov 21. Nat Med. 2017. PMID: 27869803
-
RNF43 R117fs mutant positively regulates Wnt/β-catenin signaling by failing to internalize FZD expressed on the cell surface.Sci Rep. 2022 Apr 29;12(1):7013. doi: 10.1038/s41598-022-10868-8. Sci Rep. 2022. PMID: 35487932 Free PMC article.
-
FZD5 controls intestinal crypt homeostasis and colonic Wnt surrogate agonist response.Dev Cell. 2025 Feb 3;60(3):342-351.e5. doi: 10.1016/j.devcel.2024.10.022. Epub 2024 Nov 22. Dev Cell. 2025. PMID: 39579768
-
Wnt/Frizzled signaling in hepatocellular carcinoma.Front Biosci. 2006 May 1;11:1901-15. doi: 10.2741/1933. Front Biosci. 2006. PMID: 16368566 Review.
-
Frizzled Receptors in Tumors, Focusing on Signaling, Roles, Modulation Mechanisms, and Targeted Therapies.Oncol Res. 2021 Mar 16;28(6):661-674. doi: 10.3727/096504020X16014648664459. Epub 2020 Sep 30. Oncol Res. 2021. PMID: 32998794 Free PMC article. Review.
Cited by
-
Targeting lipid metabolism: novel insights and therapeutic advances in pancreatic cancer treatment.Lipids Health Dis. 2025 Jan 13;24(1):12. doi: 10.1186/s12944-024-02426-0. Lipids Health Dis. 2025. PMID: 39806478 Free PMC article. Review.
-
Non-Coding RNAs as Critical Modulators of Cholesterol Metabolism in Cancer.Biomedicines. 2025 Jul 3;13(7):1631. doi: 10.3390/biomedicines13071631. Biomedicines. 2025. PMID: 40722703 Free PMC article. Review.
-
BAMBI Is a Prognostic Biomarker Associated with Macrophage Polarization, Glycolysis, and Lipid Metabolism in Hepatocellular Carcinoma.Int J Mol Sci. 2024 Nov 26;25(23):12713. doi: 10.3390/ijms252312713. Int J Mol Sci. 2024. PMID: 39684424 Free PMC article.
-
SUMOylation-Driven mRNA Circularization Enhances Translation and Promotes Lymphatic Metastasis of Bladder Cancer.Cancer Res. 2024 Feb 1;84(3):434-448. doi: 10.1158/0008-5472.CAN-23-2278. Cancer Res. 2024. PMID: 37991737 Free PMC article.
-
Fatty acids abrogate the growth-suppressive effects induced by inhibition of cholesterol flux in pancreatic cancer cells.Cancer Cell Int. 2023 Nov 17;23(1):276. doi: 10.1186/s12935-023-03138-8. Cancer Cell Int. 2023. PMID: 37978383 Free PMC article.
References
-
- a) Yang S., Wu Y., Xu T. H., de Waal P. W., He Y., Pu M., Chen Y., DeBruine Z. J., Zhang B., Zaidi S. A., Popov P., Guo Y., Han G. W., Lu Y., Suino‐Powell K., Dong S., Harikumar K. G., Miller L. J., Katritch V., Xu H. E., Shui W., Stevens R. C., Melcher K., Zhao S., Xu F., Nature 2018, 560, 666; - PubMed
- b) Tsutsumi N., Mukherjee S., Waghray D., Janda C. Y., Jude K. M., Miao Y., Burg J. S., Aduri N. G., Kossiakoff A. A., Gati C., Garcia K. C., Elife 2020, 9, e58464. - PMC - PubMed
-
- Xu Q., Wang Y., Dabdoub A., Smallwood P. M., Williams J., Woods C., Kelley M. W., Jiang L., Tasman W., Zhang K., Nathans J., Cell 2004, 116, 883. - PubMed
-
- a) Flanagan D. J., Phesse T. J., Barker N., Schwab R. H., Amin N., Malaterre J., Stange D. E., Nowell C. J., Currie S. A., Saw J. T., Beuchert E., Ramsay R. G., Sansom O. J., Ernst M., Clevers H., Vincan E., Stem Cell Rep. 2015, 4, 759; - PMC - PubMed
- b) Steinhart Z., Pavlovic Z., Chandrashekhar M., Hart T., Wang X., Zhang X., Robitaille M., Brown K. R., Jaksani S., Overmeer R., Boj S. F., Adams J., Pan J., Clevers H., Sidhu S., Moffat J., Angers S., Nat. Med. 2017, 23, 60; - PubMed
- c) Hao H. X., Jiang X., Cong F., Cancers 2016, 8, 54. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical