

APAF1-Binding Long Noncoding RNA Promotes Tumor Growth and Multidrug Resistance in Gastric Cancer by Blocking Apoptosome Assembly
- PMID: 35975461
- PMCID: PMC9534967
- DOI: 10.1002/advs.202201889
APAF1-Binding Long Noncoding RNA Promotes Tumor Growth and Multidrug Resistance in Gastric Cancer by Blocking Apoptosome Assembly
Abstract
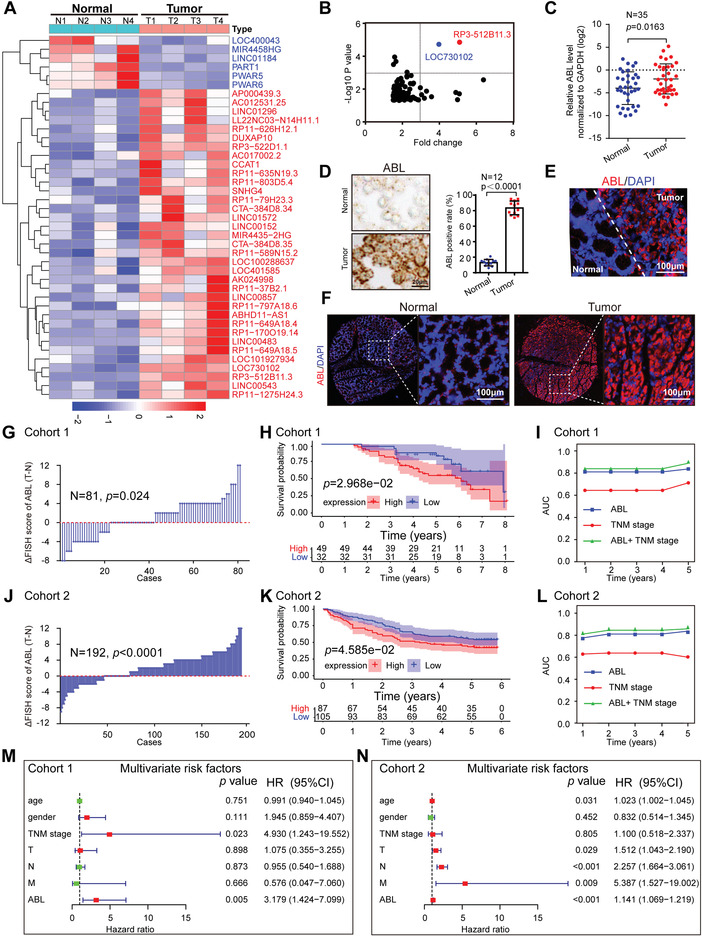
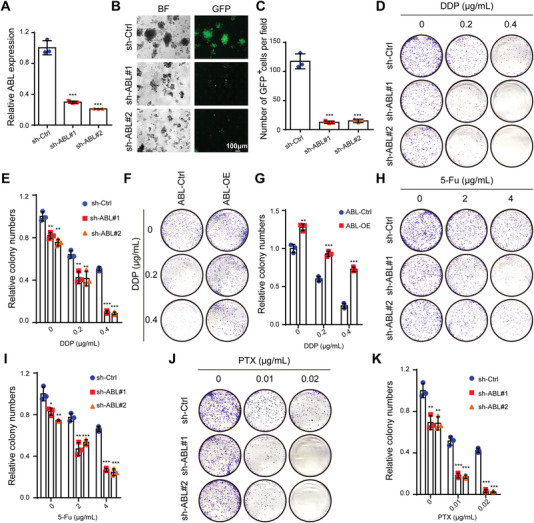
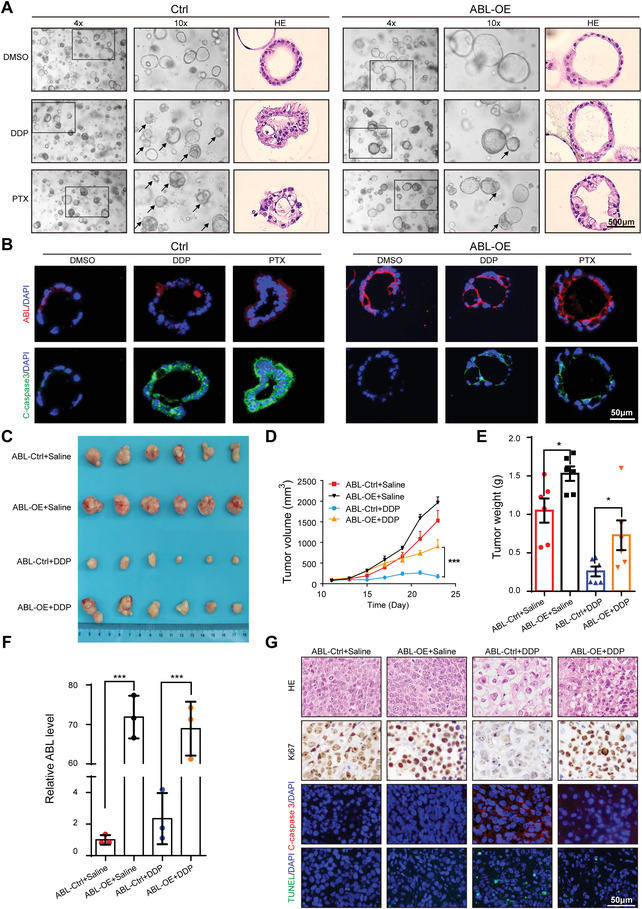
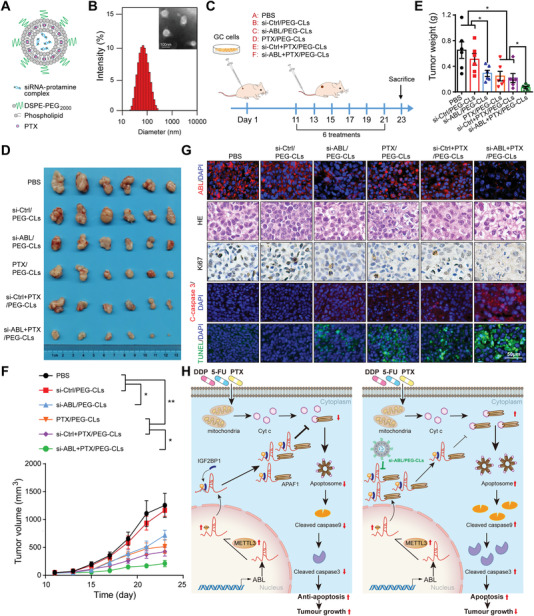
Chemotherapeutics remain the first choice for advanced gastric cancers (GCs). However, drug resistance and unavoidable severe toxicity lead to chemotherapy failure and poor prognosis. Long noncoding RNAs (lncRNAs) play critical roles in tumor progression in many cancers, including GC. Here, through RNA screening, an apoptotic protease-activating factor 1 (APAF1)-binding lncRNA (ABL) that is significantly elevated in cancerous GC tissues and an independent prognostic factor for GC patients is identified. Moreover, ABL overexpression inhibits GC cell apoptosis and promotes GC cell survival and multidrug resistance in GC xenograft and organoid models. Mechanistically, ABL directly binds to the RNA-binding protein IGF2BP1 via its KH1/2 domain, and then IGF2BP1 further recognizes the METTL3-mediated m6A modification on ABL, which maintains ABL stability. In addition, ABL can bind to the WD1/WD2 domain of APAF1, which competitively prevent cytochrome c from interacting with APAF1, blocking apoptosome assembly and caspase-9/3 activation; these events lead to resistance to cell death in GC cells. Intriguingly, targeting ABL using encapsulated liposomal siRNA can significantly enhance the sensitivity of GC cells to chemotherapy. Collectively, the results suggest that ABL can be a potential prognostic biomarker and therapeutic target in GC.
Keywords: ABL; IGF2BP1; apoptosis; apoptotic protease-activating factor 1 (APAF1); drug resistance; gastric cancer; m6A.
© 2022 The Authors. Advanced Science published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



References
-
- a) Chen W., Zheng R., Baade P. D., Zhang S., Zeng H., Bray F., Jemal A., Yu X. Q., He J., Ca‐Cancer J. Clin. 2016, 66, 115; - PubMed
- b) Ferlay J., Colombet M., Soerjomataram I., Mathers C., Parkin D. M., Pineros M., Znaor A., Bray F., Int. J. Cancer 2019, 144, 1941; - PubMed
- c) Smyth E. C., Nilsson M., Grabsch H. I., van Grieken N. C., Lordick F., Lancet 2020, 396, 635. - PubMed
-
- Wang Q., Xiong J., Qiu D., Zhao X., Yan D., Xu W., Wang Z., Chen Q., Panday S., Li A., Wang S., Zhou J., Int. J. Biochem. Cell Biol. 2017, 92, 164. - PubMed
-
- a) Kang Y. K., Chin K., Chung H. C., Kadowaki S., Oh S. C., Nakayama N., Lee K. W., Hara H., Chung I. J., Tsuda M., Park S. H., Hosaka H., Hironaka S., Miyata Y., Ryu M. H., Baba H., Hyodo I., Bang Y. J., Boku N., Lancet 2020, 21, 1045; - PubMed
- b) Shitara K., Van Cutsem E., Bang Y. J., Fuchs C., Wyrwicz L., Lee K. W., Kudaba I., Garrido M., Chung H. C., Lee J., Castro H. R., Mansoor W., Braghiroli M. I., Karaseva N., Caglevic C., Villanueva L., Goekkurt E., Satake H., Enzinger P., Alsina M., Benson A., Chao J., Ko A. H., Wainberg Z. A., Kher U., Shah S., Kang S. P., Tabernero J., JAMA, J. Am. Med. Assoc. Oncol. 2020, 6, 1571; - PMC - PubMed
- c) Hong C., Wang D., Liang J., Guo Y., Zhu Y., Xia J., Qin J., Zhan H., Wang J., Theranostics 2019, 9, 4437. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 82073114/National Natural Science Foundation of China
- 81903085/National Natural Science Foundation of China
- 81773383/National Natural Science Foundation of China
- 82102984/National Natural Science Foundation of China
- 82103266/National Natural Science Foundation of China
- 2020T130291/China Postdoctoral Science Foundation
- 2019M651808/China Postdoctoral Science Foundation
- 2020M681562/China Postdoctoral Science Foundation
- BK20170047/Science Foundation for Distinguished Young Scholars of Jiangsu Province
- 021414380439/Fundamental Research Funds for the Central Universities
- JQX21005/Nanjing special foundation for health science and technology development distinguished young program
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous