

Moxifloxacin-Mediated Killing of Mycobacterium tuberculosis Involves Respiratory Downshift, Reductive Stress, and Accumulation of Reactive Oxygen Species
- PMID: 35975988
- PMCID: PMC9487606
- DOI: 10.1128/aac.00592-22
Moxifloxacin-Mediated Killing of Mycobacterium tuberculosis Involves Respiratory Downshift, Reductive Stress, and Accumulation of Reactive Oxygen Species
Abstract
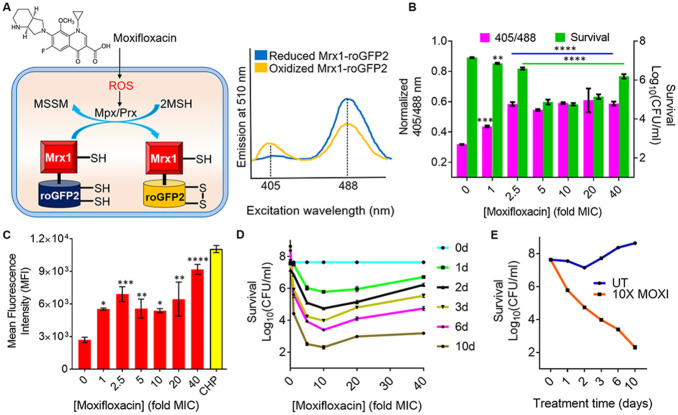
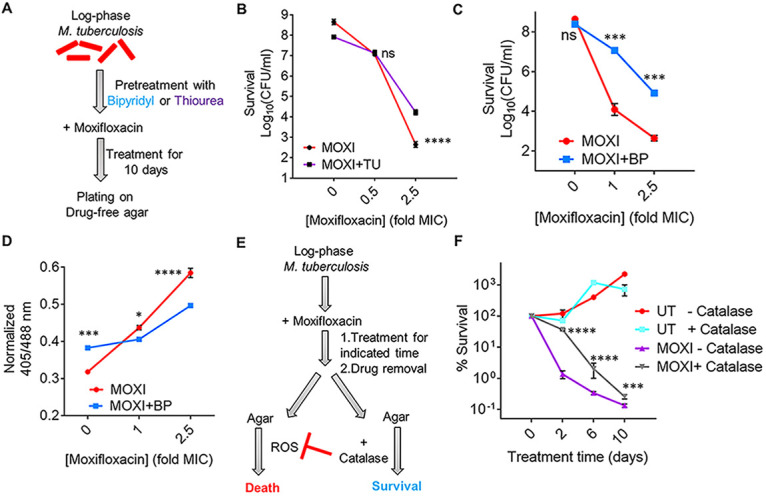
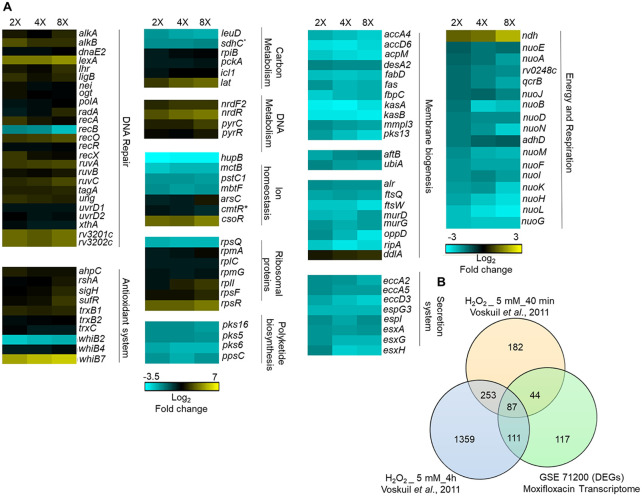
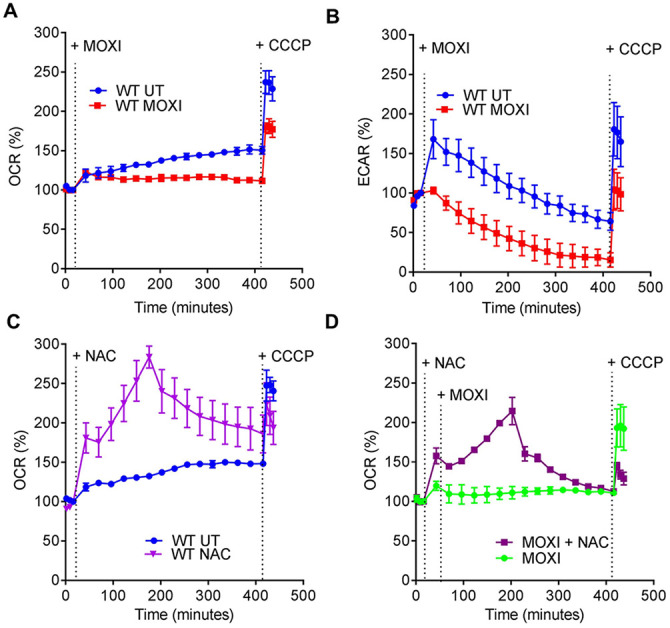
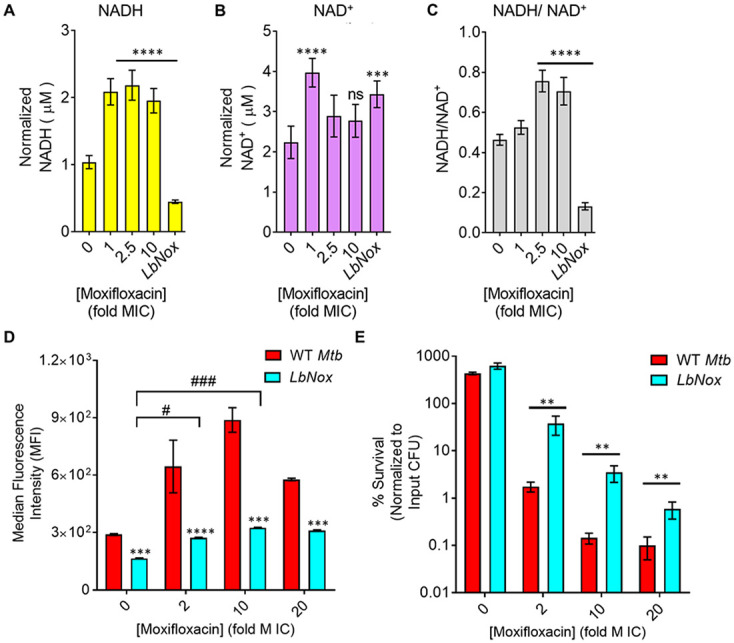
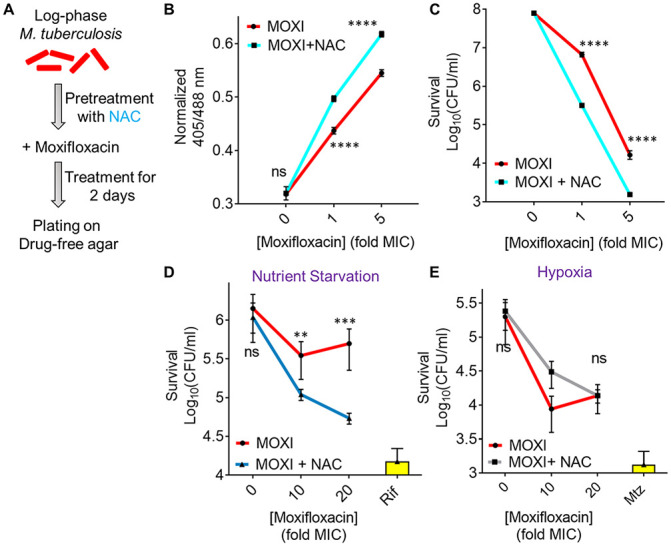
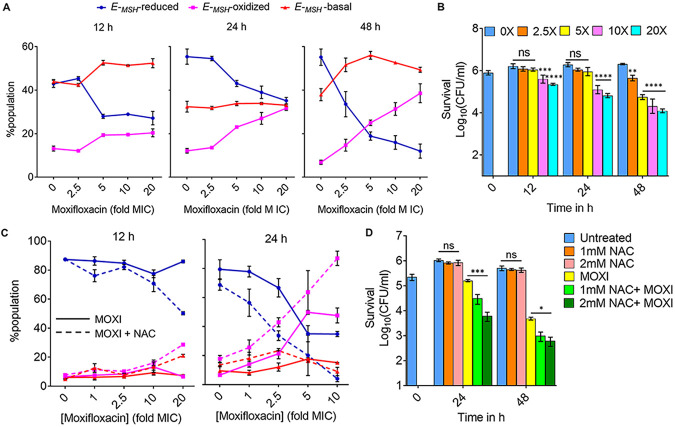
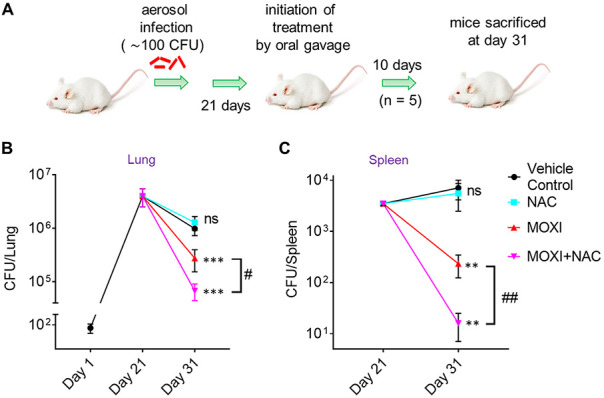
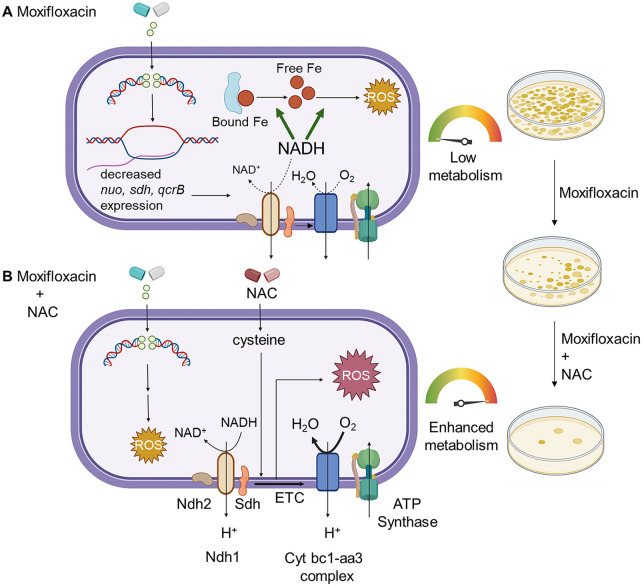
Moxifloxacin is central to treatment of multidrug-resistant tuberculosis. Effects of moxifloxacin on the Mycobacterium tuberculosis redox state were explored to identify strategies for increasing lethality and reducing the prevalence of extensively resistant tuberculosis. A noninvasive redox biosensor and a reactive oxygen species (ROS)-sensitive dye revealed that moxifloxacin induces oxidative stress correlated with M. tuberculosis death. Moxifloxacin lethality was mitigated by supplementing bacterial cultures with an ROS scavenger (thiourea), an iron chelator (bipyridyl), and, after drug removal, an antioxidant enzyme (catalase). Lethality was also reduced by hypoxia and nutrient starvation. Moxifloxacin increased the expression of genes involved in the oxidative stress response, iron-sulfur cluster biogenesis, and DNA repair. Surprisingly, and in contrast with Escherichia coli studies, moxifloxacin decreased expression of genes involved in respiration, suppressed oxygen consumption, increased the NADH/NAD+ ratio, and increased the labile iron pool in M. tuberculosis. Lowering the NADH/NAD+ ratio in M. tuberculosis revealed that NADH-reductive stress facilitates an iron-mediated ROS surge and moxifloxacin lethality. Treatment with N-acetyl cysteine (NAC) accelerated respiration and ROS production, increased moxifloxacin lethality, and lowered the mutant prevention concentration. Moxifloxacin induced redox stress in M. tuberculosis inside macrophages, and cotreatment with NAC potentiated the antimycobacterial efficacy of moxifloxacin during nutrient starvation, inside macrophages, and in mice, where NAC restricted the emergence of resistance. Thus, NADH-reductive stress contributes to moxifloxacin-mediated killing of M. tuberculosis, and the respiration stimulator (NAC) enhances lethality and suppresses the emergence of drug resistance.
Keywords: N-acetyl cysteine; NADH; ROS; antimycobacterial; fluoroquinolone; moxifloxacin; oxidative stress; redox biosensor; reductive stress; resistance; respiration.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Fluoroquinolone heteroresistance, antimicrobial tolerance, and lethality enhancement.Front Cell Infect Microbiol. 2022 Sep 29;12:938032. doi: 10.3389/fcimb.2022.938032. eCollection 2022. Front Cell Infect Microbiol. 2022. PMID: 36250047 Free PMC article. Review.
-
Contribution of reactive oxygen species to pathways of quinolone-mediated bacterial cell death.J Antimicrob Chemother. 2010 Mar;65(3):520-4. doi: 10.1093/jac/dkp486. Epub 2010 Jan 12. J Antimicrob Chemother. 2010. PMID: 20067982 Free PMC article.
-
Inhibitors of reactive oxygen species accumulation delay and/or reduce the lethality of several antistaphylococcal agents.Antimicrob Agents Chemother. 2012 Nov;56(11):6048-50. doi: 10.1128/AAC.00754-12. Epub 2012 Sep 4. Antimicrob Agents Chemother. 2012. PMID: 22948880 Free PMC article.
-
N-acetyl-cysteine exhibits potent anti-mycobacterial activity in addition to its known anti-oxidative functions.BMC Microbiol. 2016 Oct 28;16(1):251. doi: 10.1186/s12866-016-0872-7. BMC Microbiol. 2016. PMID: 27793104 Free PMC article.
-
Transcriptional regulation of bacterial virulence gene expression by molecular oxygen and nitric oxide.Virulence. 2014;5(8):794-809. doi: 10.4161/viru.27794. Epub 2014 Oct 31. Virulence. 2014. PMID: 25603427 Free PMC article. Review.
Cited by
-
Quorum-sensing agr system of Staphylococcus aureus primes gene expression for protection from lethal oxidative stress.Elife. 2024 Apr 30;12:RP89098. doi: 10.7554/eLife.89098. Elife. 2024. PMID: 38687677 Free PMC article.
-
Combination of gallium citrate and levofloxacin induces a distinct metabolome profile and enhances growth inhibition of multidrug-resistant Mycobacterium tuberculosis compared to linezolid.Front Microbiol. 2024 Nov 29;15:1474071. doi: 10.3389/fmicb.2024.1474071. eCollection 2024. Front Microbiol. 2024. PMID: 39697659 Free PMC article.
-
Antibiotic-persistent bacterial cells exhibiting low-level ROS are eradicated by ROS-independent membrane disruption.mBio. 2025 Aug 13;16(8):e0119925. doi: 10.1128/mbio.01199-25. Epub 2025 Jun 30. mBio. 2025. PMID: 40586599 Free PMC article.
-
Mycobacterium tuberculosis Rv1048c affects the biological characteristics of recombinant Mycobacterium smegmatis.Sci Rep. 2024 Nov 29;14(1):29749. doi: 10.1038/s41598-024-81405-y. Sci Rep. 2024. PMID: 39613837 Free PMC article.
-
Incorporation of macrophage immune stresses into an assay for drug tolerance in intracellular Mycobacterium tuberculosis.bioRxiv [Preprint]. 2025 May 9:2025.05.09.653069. doi: 10.1101/2025.05.09.653069. bioRxiv. 2025. PMID: 40655004 Free PMC article. Preprint.
References
-
- WHO. 2019. Global tuberculosis report 2019. World Health Organization, Geneva, Switzerland.
-
- WHO. 2013. Global tuberculosis report 2013. World Health Organization, Geneva, Switzerland.
-
- Shandil RK, Jayaram R, Kaur P, Gaonkar S, Suresh BL, Mahesh BN, Jayashree R, Nandi V, Bharath S, Balasubramanian V. 2007. Moxifloxacin, ofloxacin, sparfloxacin, and ciprofloxacin against Mycobacterium tuberculosis: evaluation of in vitro and pharmacodynamic indices that best predict in vivo efficacy. Antimicrob Agents Chemother 51:576–582. 10.1128/AAC.00414-06. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
