

AlphaFold, Artificial Intelligence (AI), and Allostery
- PMID: 35976160
- PMCID: PMC9442638
- DOI: 10.1021/acs.jpcb.2c04346
AlphaFold, Artificial Intelligence (AI), and Allostery
Abstract
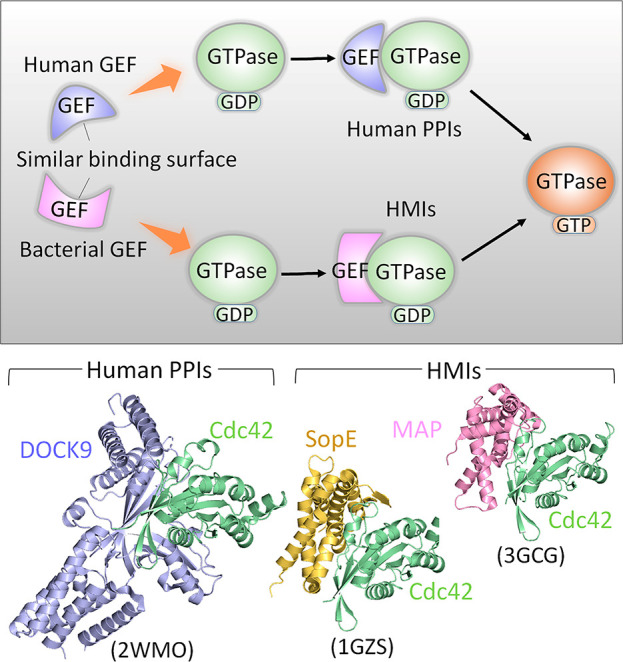
AlphaFold has burst into our lives. A powerful algorithm that underscores the strength of biological sequence data and artificial intelligence (AI). AlphaFold has appended projects and research directions. The database it has been creating promises an untold number of applications with vast potential impacts that are still difficult to surmise. AI approaches can revolutionize personalized treatments and usher in better-informed clinical trials. They promise to make giant leaps toward reshaping and revamping drug discovery strategies, selecting and prioritizing combinations of drug targets. Here, we briefly overview AI in structural biology, including in molecular dynamics simulations and prediction of microbiota-human protein-protein interactions. We highlight the advancements accomplished by the deep-learning-powered AlphaFold in protein structure prediction and their powerful impact on the life sciences. At the same time, AlphaFold does not resolve the decades-long protein folding challenge, nor does it identify the folding pathways. The models that AlphaFold provides do not capture conformational mechanisms like frustration and allostery, which are rooted in ensembles, and controlled by their dynamic distributions. Allostery and signaling are properties of populations. AlphaFold also does not generate ensembles of intrinsically disordered proteins and regions, instead describing them by their low structural probabilities. Since AlphaFold generates single ranked structures, rather than conformational ensembles, it cannot elucidate the mechanisms of allosteric activating driver hotspot mutations nor of allosteric drug resistance. However, by capturing key features, deep learning techniques can use the single predicted conformation as the basis for generating a diverse ensemble.
Conflict of interest statement
The authors declare no competing financial interest.
Figures


References
-
- Varadi M.; Anyango S.; Deshpande M.; Nair S.; Natassia C.; Yordanova G.; Yuan D.; Stroe O.; Wood G.; Laydon A.; et al. AlphaFold protein structure database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50 (D1), D439–D444. 10.1093/nar/gkab1061. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
