

Live-seq enables temporal transcriptomic recording of single cells
- PMID: 35978187
- PMCID: PMC9402441
- DOI: 10.1038/s41586-022-05046-9
Live-seq enables temporal transcriptomic recording of single cells
Abstract
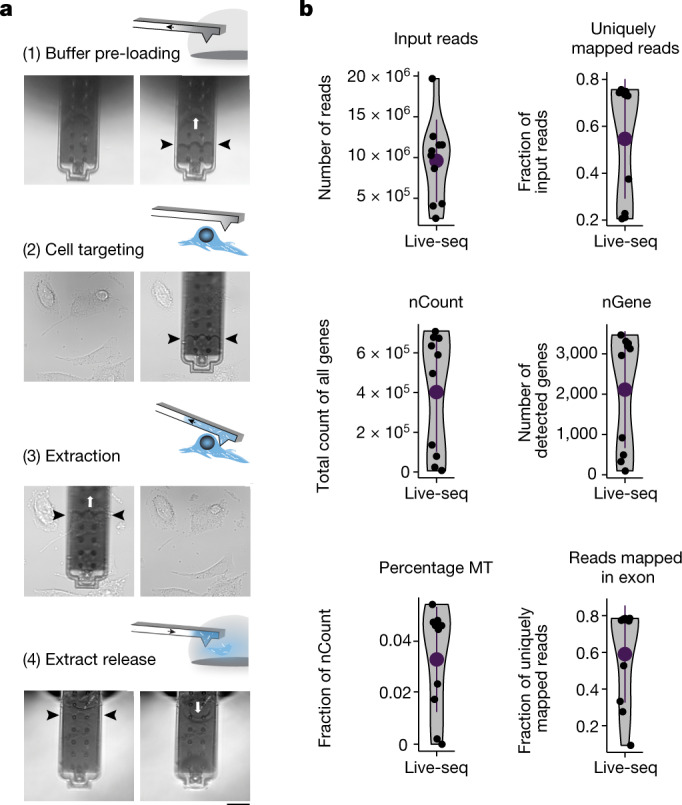
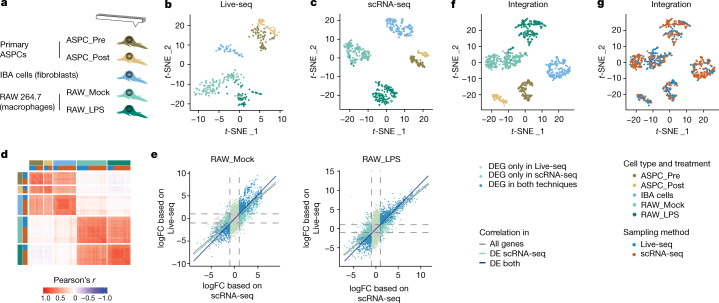
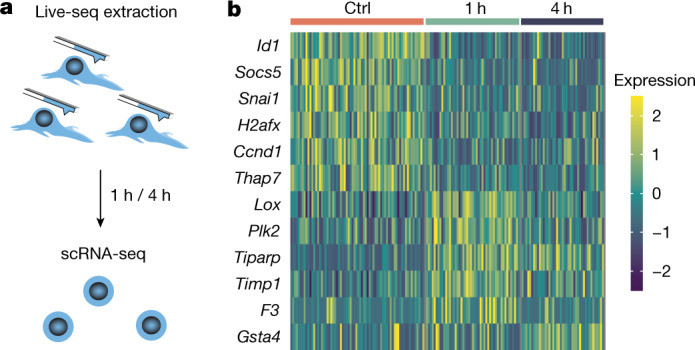
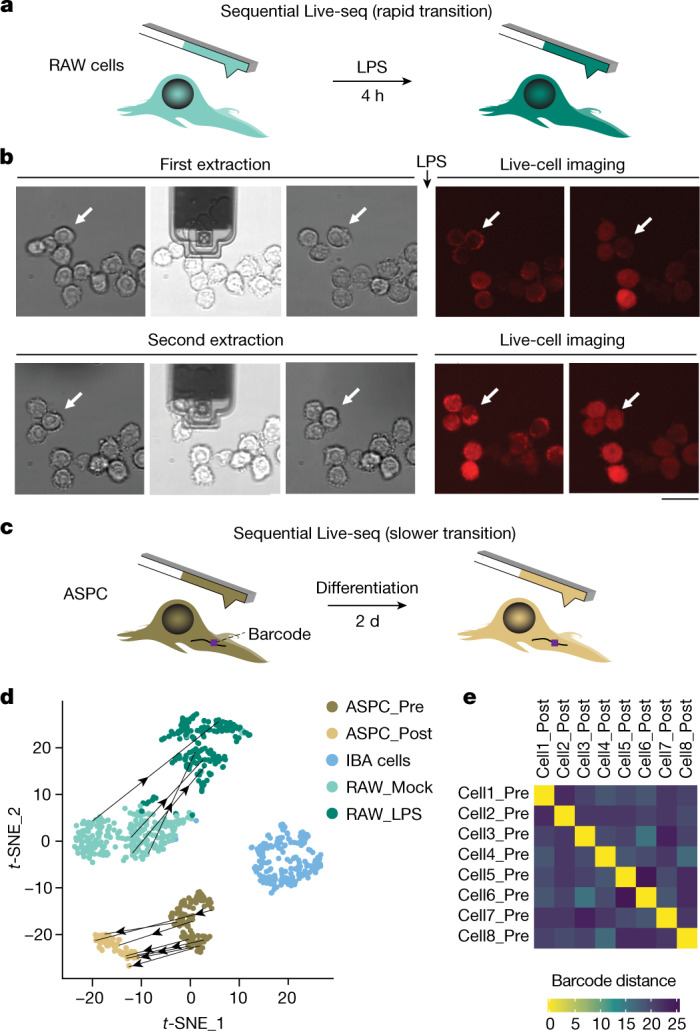
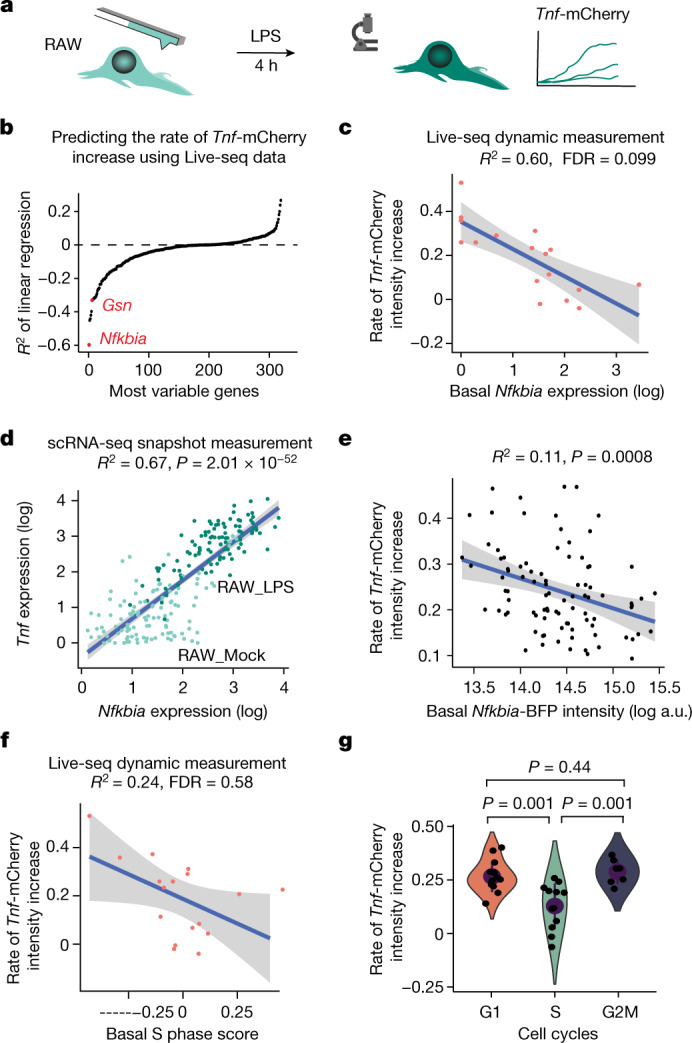
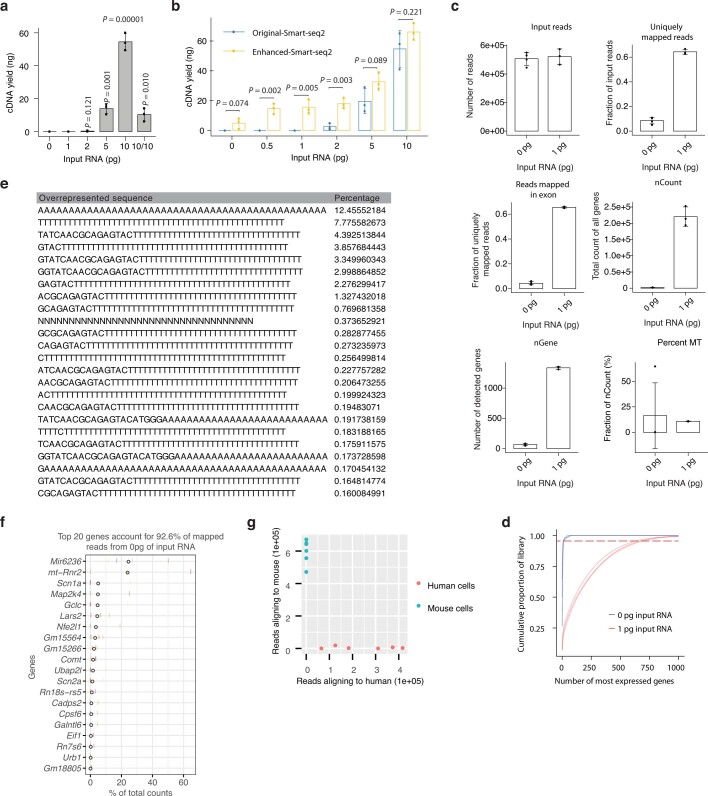
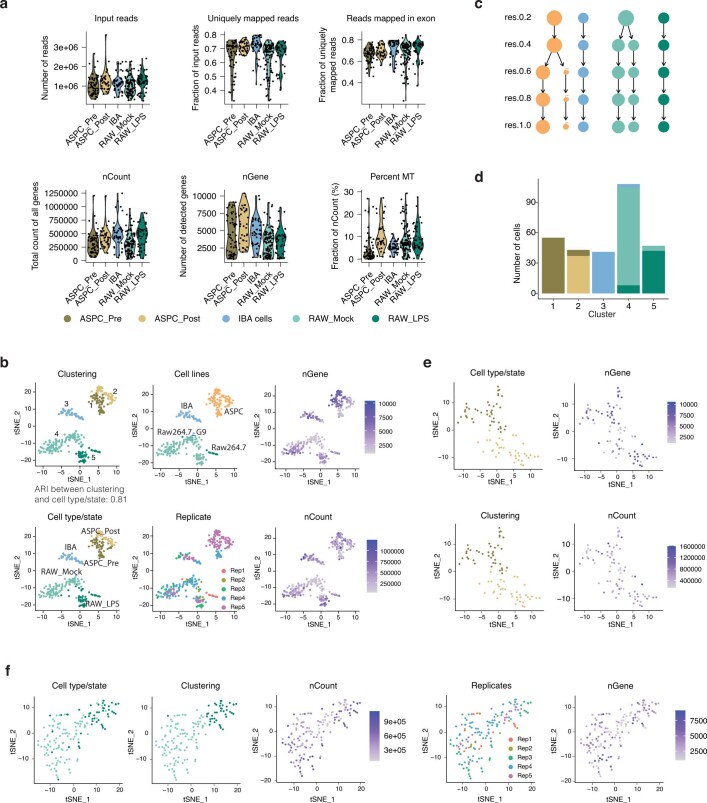
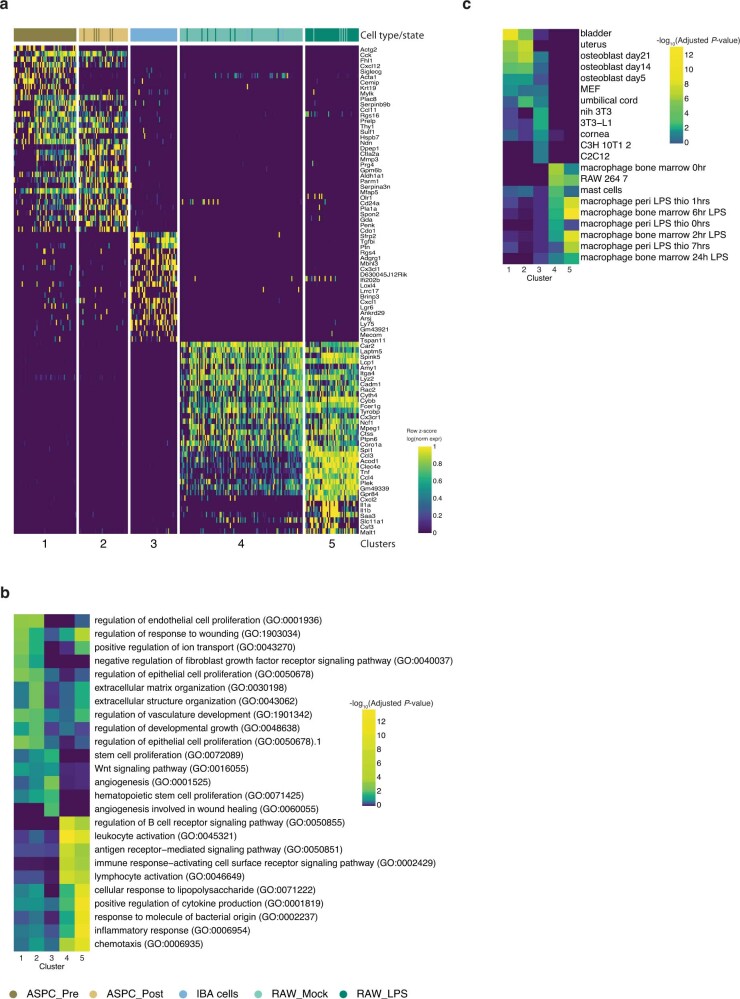
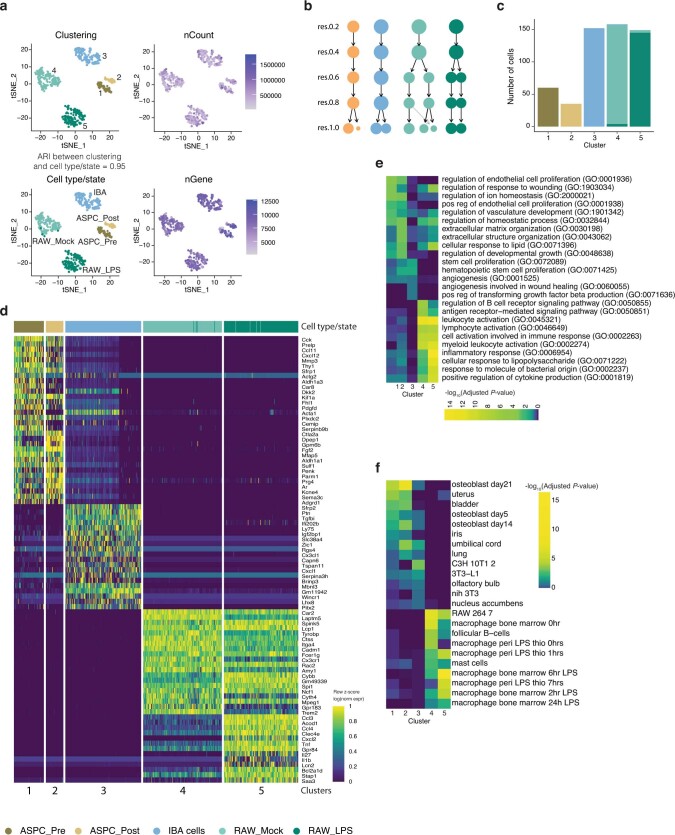
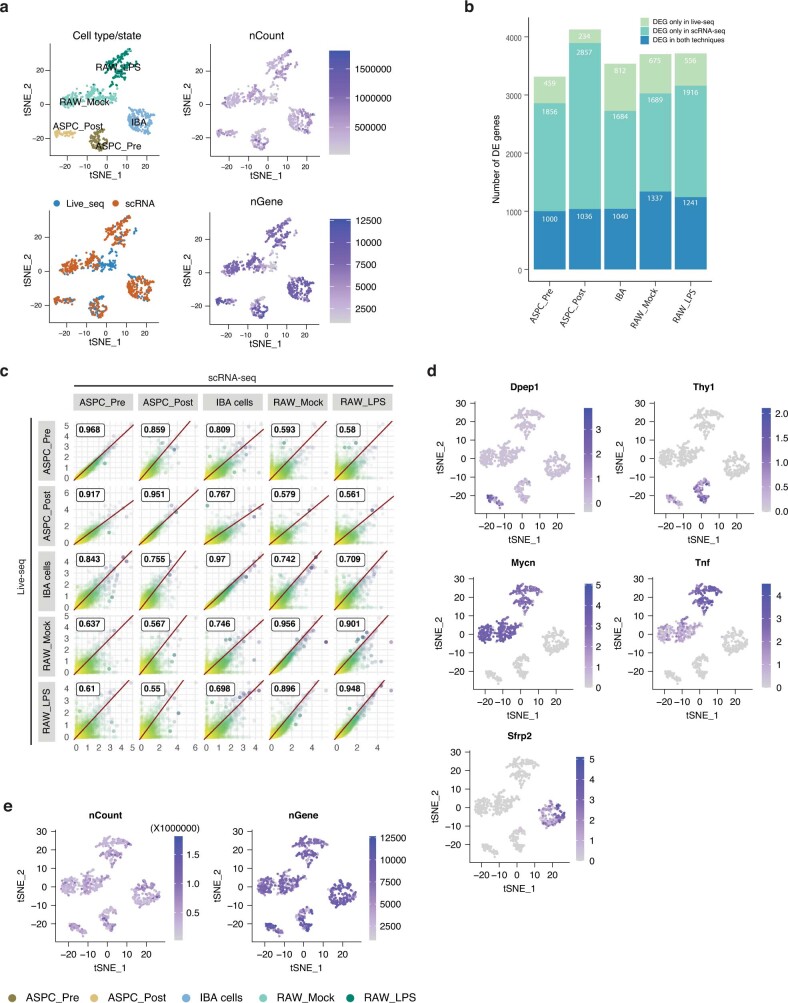
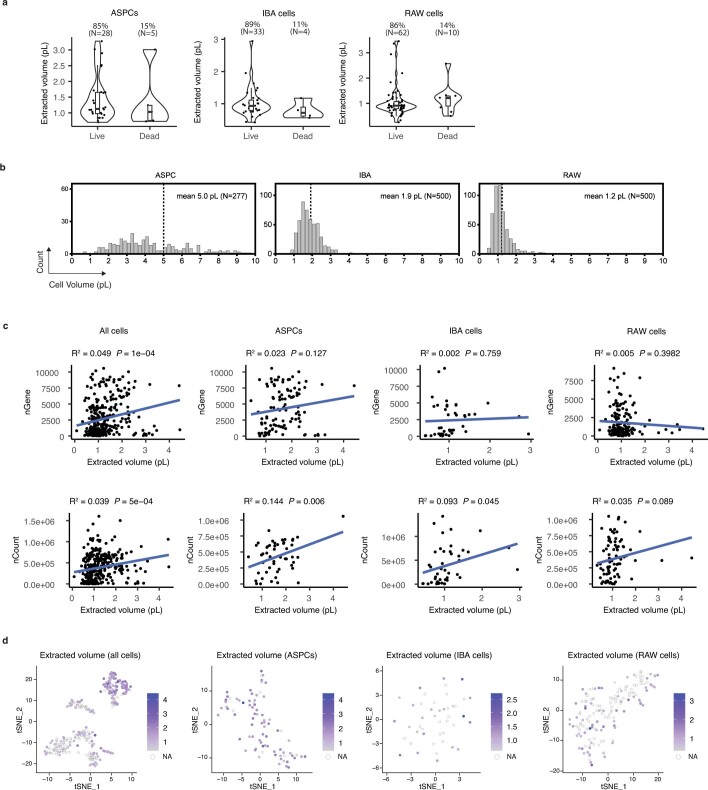
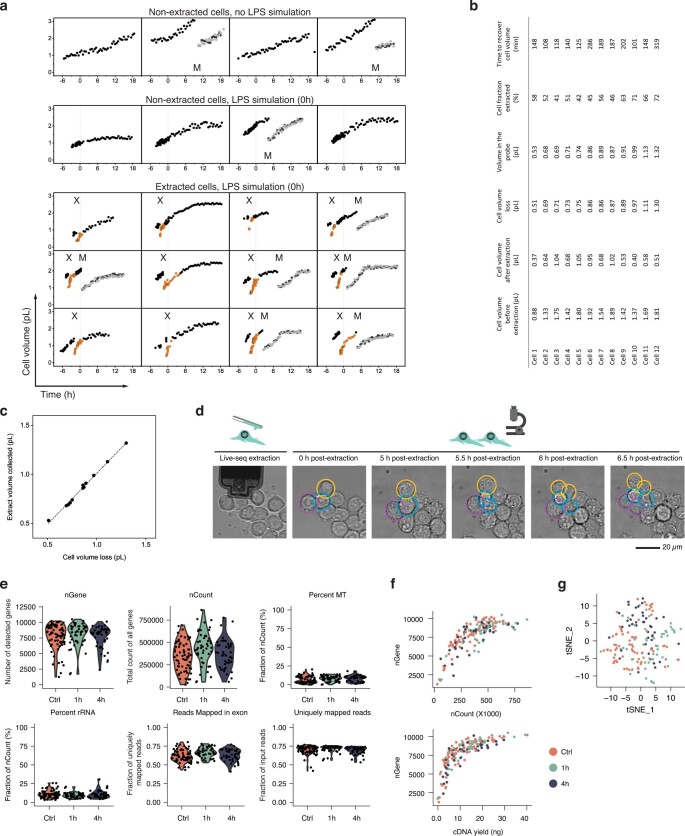
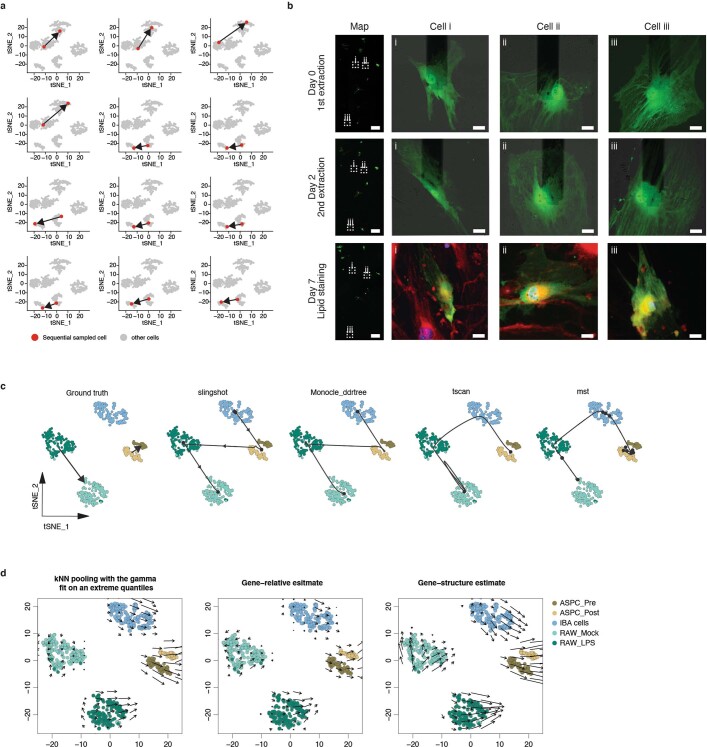
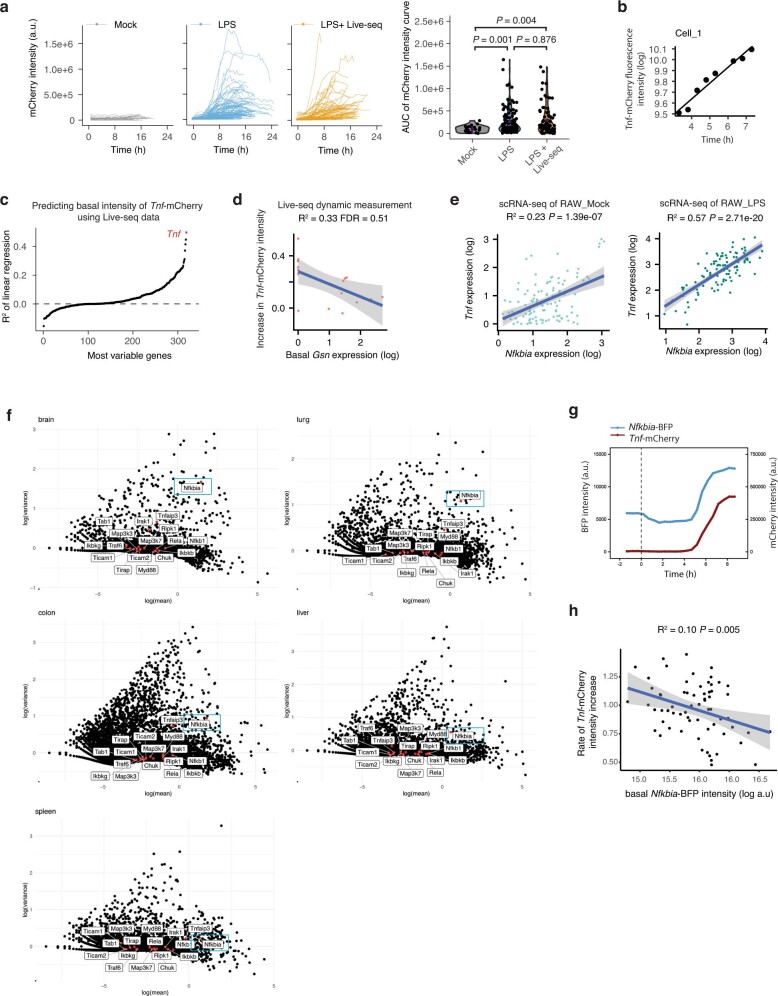
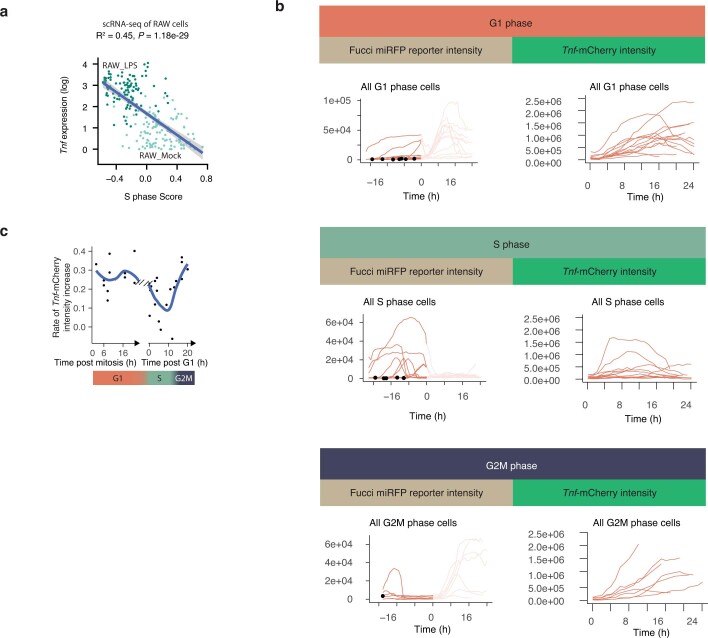
Single-cell transcriptomics (scRNA-seq) has greatly advanced our ability to characterize cellular heterogeneity1. However, scRNA-seq requires lysing cells, which impedes further molecular or functional analyses on the same cells. Here, we established Live-seq, a single-cell transcriptome profiling approach that preserves cell viability during RNA extraction using fluidic force microscopy2,3, thus allowing to couple a cell's ground-state transcriptome to its downstream molecular or phenotypic behaviour. To benchmark Live-seq, we used cell growth, functional responses and whole-cell transcriptome read-outs to demonstrate that Live-seq can accurately stratify diverse cell types and states without inducing major cellular perturbations. As a proof of concept, we show that Live-seq can be used to directly map a cell's trajectory by sequentially profiling the transcriptomes of individual macrophages before and after lipopolysaccharide (LPS) stimulation, and of adipose stromal cells pre- and post-differentiation. In addition, we demonstrate that Live-seq can function as a transcriptomic recorder by preregistering the transcriptomes of individual macrophages that were subsequently monitored by time-lapse imaging after LPS exposure. This enabled the unsupervised, genome-wide ranking of genes on the basis of their ability to affect macrophage LPS response heterogeneity, revealing basal Nfkbia expression level and cell cycle state as important phenotypic determinants, which we experimentally validated. Thus, Live-seq can address a broad range of biological questions by transforming scRNA-seq from an end-point to a temporal analysis approach.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Single-cell RNA-seq keeps cells alive.Nat Biotechnol. 2022 Oct;40(10):1432. doi: 10.1038/s41587-022-01515-8. Nat Biotechnol. 2022. PMID: 36207601 No abstract available.
-
Single-cell temporal transcriptomics from tiny cytoplasmic biopsies.Cell Rep Methods. 2022 Oct 17;2(10):100319. doi: 10.1016/j.crmeth.2022.100319. eCollection 2022 Oct 24. Cell Rep Methods. 2022. PMID: 36313799 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
