

Sporadic Creutzfeldt-Jakob disease VM1: phenotypic and molecular characterization of a novel subtype of human prion disease
- PMID: 35978418
- PMCID: PMC9387077
- DOI: 10.1186/s40478-022-01415-7
Sporadic Creutzfeldt-Jakob disease VM1: phenotypic and molecular characterization of a novel subtype of human prion disease
Abstract
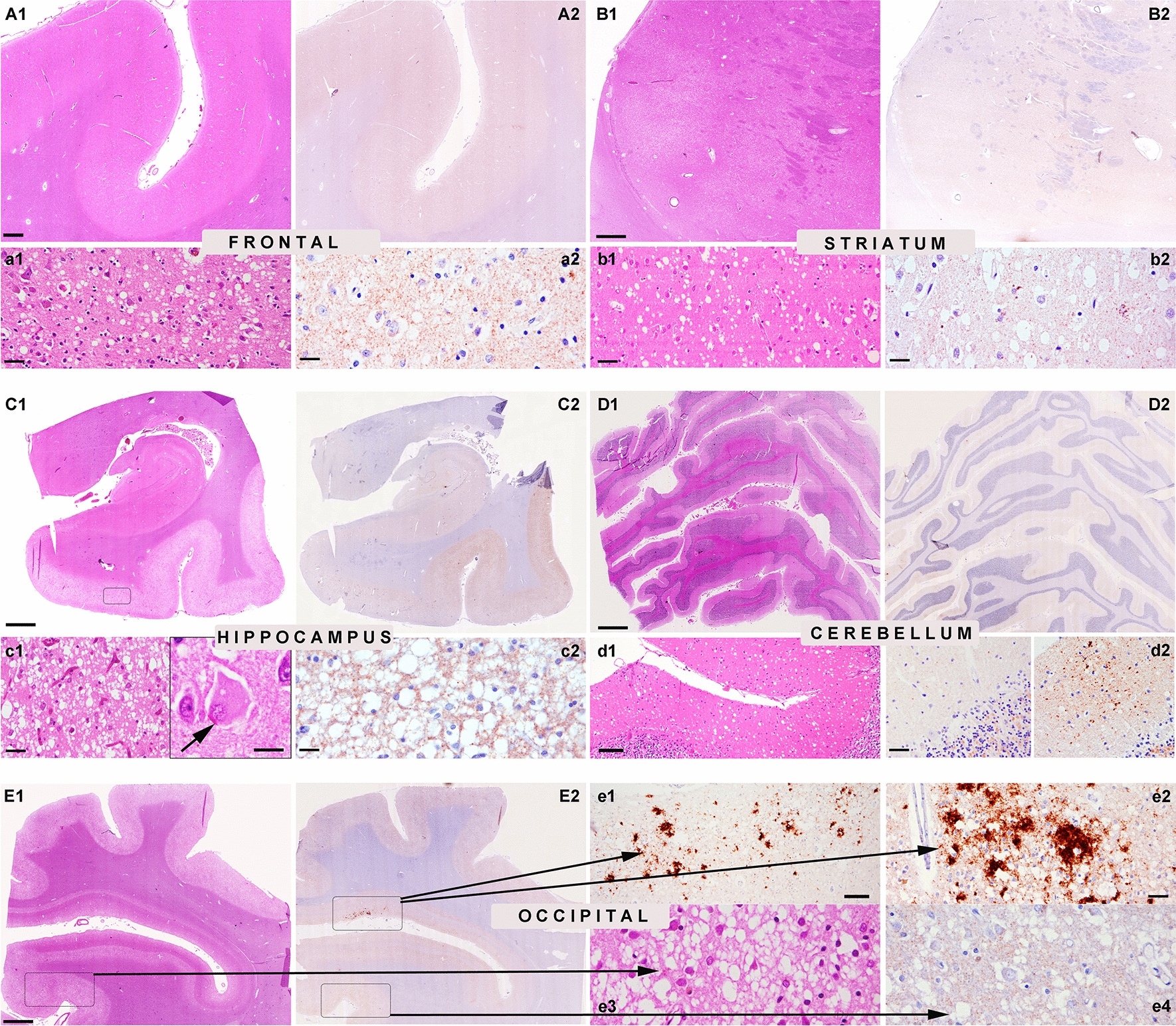
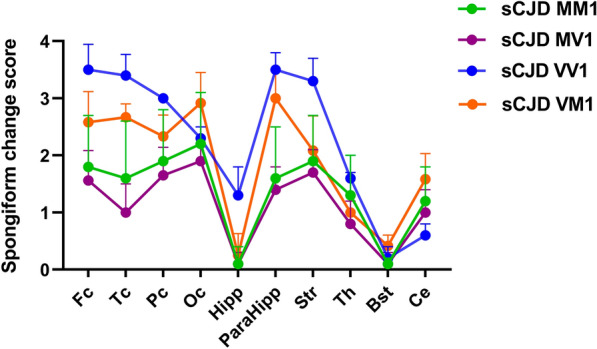
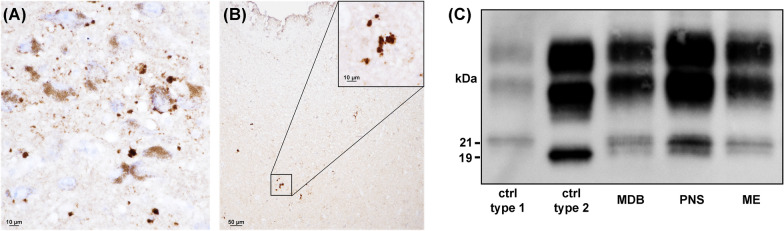
The methionine (M)-valine (V) polymorphic codon 129 of the prion protein gene (PRNP) plays a central role in both susceptibility and phenotypic expression of sporadic Creutzfeldt-Jakob diseases (sCJD). Experimental transmissions of sCJD in humanized transgenic mice led to the isolation of five prion strains, named M1, M2C, M2T, V2, and V1, based on two major conformations of the pathological prion protein (PrPSc, type 1 and type 2), and the codon 129 genotype determining susceptibility and propagation efficiency. While the most frequent sCJD strains have been described in codon 129 homozygosis (MM1, MM2C, VV2) and heterozygosis (MV1, MV2K, and MV2C), the V1 strain has only been found in patients carrying VV. We identified six sCJD cases, 4 in Catalonia and 2 in Italy, carrying MV at PRNP codon 129 in combination with PrPSc type 1 and a new clinical and neuropathological profile reminiscent of the VV1 sCJD subtype rather than typical MM1/MV1. All patients had a relatively long duration (mean of 20.5 vs. 3.5 months of MM1/MV1 patients) and lacked electroencephalographic periodic sharp-wave complexes at diagnosis. Distinctive histopathological features included the spongiform change with vacuoles of larger size than those seen in sCJD MM1/MV1, the lesion profile with prominent cortical and striatal involvement, and the pattern of PrPSc deposition characterized by a dissociation between florid spongiform change and mild synaptic deposits associated with coarse, patch-like deposits in the cerebellar molecular layer. Western blot analysis of brain homogenates revealed a PrPSc type 1 profile with physicochemical properties reminiscent of the type 1 protein linked to the VV1 sCJD subtype. In summary, we have identified a new subtype of sCJD with distinctive clinicopathological features significantly overlapping with those of the VV1 subtype, possibly representing the missing evidence of V1 sCJD strain propagation in the 129MV host genotype.
Keywords: CJD; Classification; Codon 129; Histotype; PRNP; PrP; Prion disease; Prion strains.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

Similar articles
-
Prions from Sporadic Creutzfeldt-Jakob Disease Patients Propagate as Strain Mixtures.mBio. 2020 Jun 16;11(3):e00393-20. doi: 10.1128/mBio.00393-20. mBio. 2020. PMID: 32546613 Free PMC article.
-
Phenotypic diversity of genetic Creutzfeldt-Jakob disease: a histo-molecular-based classification.Acta Neuropathol. 2021 Oct;142(4):707-728. doi: 10.1007/s00401-021-02350-y. Epub 2021 Jul 29. Acta Neuropathol. 2021. PMID: 34324063 Free PMC article.
-
Prion Strain Characterization of a Novel Subtype of Creutzfeldt-Jakob Disease.J Virol. 2017 May 12;91(11):e02390-16. doi: 10.1128/JVI.02390-16. Print 2017 Jun 1. J Virol. 2017. PMID: 28298604 Free PMC article.
-
Pathological spectrum of sporadic Creutzfeldt-Jakob disease.Pathology. 2025 Mar;57(2):196-206. doi: 10.1016/j.pathol.2024.09.005. Epub 2024 Nov 13. Pathology. 2025. PMID: 39665904 Review.
-
Creutzfeldt-Jakob disease.Adv Exp Med Biol. 2012;724:76-90. doi: 10.1007/978-1-4614-0653-2_6. Adv Exp Med Biol. 2012. PMID: 22411235 Review.
Cited by
-
Case report: Two clusters of Creutzfeldt-Jakob disease cases within 1 year in West Michigan.Front Neurol. 2023 Mar 20;14:1134225. doi: 10.3389/fneur.2023.1134225. eCollection 2023. Front Neurol. 2023. PMID: 37021286 Free PMC article.
-
A novel subtype of sporadic Creutzfeldt-Jakob disease with PRNP codon 129MM genotype and PrP plaques.Acta Neuropathol. 2023 Jul;146(1):121-143. doi: 10.1007/s00401-023-02581-1. Epub 2023 May 8. Acta Neuropathol. 2023. PMID: 37156880 Free PMC article.
-
Mood Alterations in the Prodromal Phase of Sporadic Creutzfeldt-Jakob Disease.JAMA Neurol. 2025 Feb 1;82(2):185-192. doi: 10.1001/jamaneurol.2024.4447. JAMA Neurol. 2025. PMID: 39786417
-
The Zoonotic Potential of Chronic Wasting Disease-A Review.Foods. 2023 Feb 15;12(4):824. doi: 10.3390/foods12040824. Foods. 2023. PMID: 36832899 Free PMC article. Review.
-
Novel histotypes of sporadic Creutzfeldt-Jakob disease linked to 129MV genotype.Acta Neuropathol Commun. 2023 Aug 31;11(1):141. doi: 10.1186/s40478-023-01631-9. Acta Neuropathol Commun. 2023. PMID: 37653534 Free PMC article.
References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Research Materials
