

Metagenomics versus total RNA sequencing: most accurate data-processing tools, microbial identification accuracy and perspectives for ecological assessments
- PMID: 35979944
- PMCID: PMC9458450
- DOI: 10.1093/nar/gkac689
Metagenomics versus total RNA sequencing: most accurate data-processing tools, microbial identification accuracy and perspectives for ecological assessments
Abstract
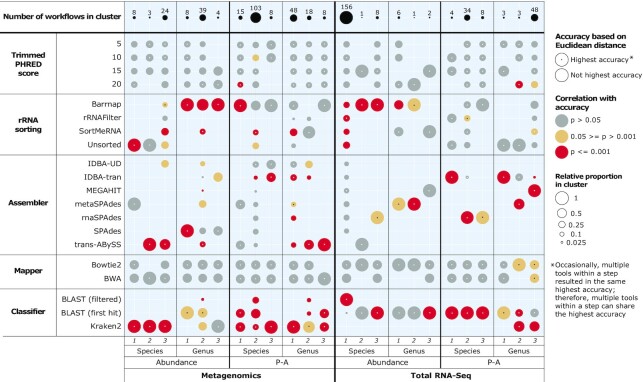
Metagenomics and total RNA sequencing (total RNA-Seq) have the potential to improve the taxonomic identification of diverse microbial communities, which could allow for the incorporation of microbes into routine ecological assessments. However, these target-PCR-free techniques require more testing and optimization. In this study, we processed metagenomics and total RNA-Seq data from a commercially available microbial mock community using 672 data-processing workflows, identified the most accurate data-processing tools, and compared their microbial identification accuracy at equal and increasing sequencing depths. The accuracy of data-processing tools substantially varied among replicates. Total RNA-Seq was more accurate than metagenomics at equal sequencing depths and even at sequencing depths almost one order of magnitude lower than those of metagenomics. We show that while data-processing tools require further exploration, total RNA-Seq might be a favorable alternative to metagenomics for target-PCR-free taxonomic identifications of microbial communities and might enable a substantial reduction in sequencing costs while maintaining accuracy. This could be particularly an advantage for routine ecological assessments, which require cost-effective yet accurate methods, and might allow for the incorporation of microbes into ecological assessments.
© The Author(s) 2022. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures




References
-
- IPBES Services Díaz S., Settele J., Brondízio E.S., Ngo H.T., Guèze M., Agard J., Razzaque J., Arneth A., Balvanera P., Brauman K.A.et al... Summary for Policymakers of the Global Assessment Report on Biodiversity and Ecosystem Services of the Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem. 2019; Bonn, Germany.
-
- WWF Almond R.E.A., Grooten M., Petersen T.. Living Planet Report 2020 - Bending the Curve of Biodiversity Loss. 2020; Gland, Switzerland.
-
- Pettorelli N., Graham N.A.J., Seddon N., Maria da Cunha Bustamante M., Lowton M.J., Sutherland W.J., Koldewey H.J., Prentice H.C., Barlow J.. Time to integrate global climate change and biodiversity science-policy agendas. J. Appl. Ecol. 2021; 58:2384–2393.
-
- Kubiszewski I., Costanza R., Anderson S., Sutton P.. The future value of ecosystem services: global scenarios and national implications. Ecosyst. Serv. 2017; 26:289–301.
-
- Burger J. Bioindicators: a review of their use in the environmental literature 1970–2005. Environ. Bioindic. 2006; 1:136–144.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
