

Recovering individual haplotypes and a contiguous genome assembly from pooled long-read sequencing of the diamondback moth (Lepidoptera: Plutellidae)
- PMID: 35980174
- PMCID: PMC9526047
- DOI: 10.1093/g3journal/jkac210
Recovering individual haplotypes and a contiguous genome assembly from pooled long-read sequencing of the diamondback moth (Lepidoptera: Plutellidae)
Abstract
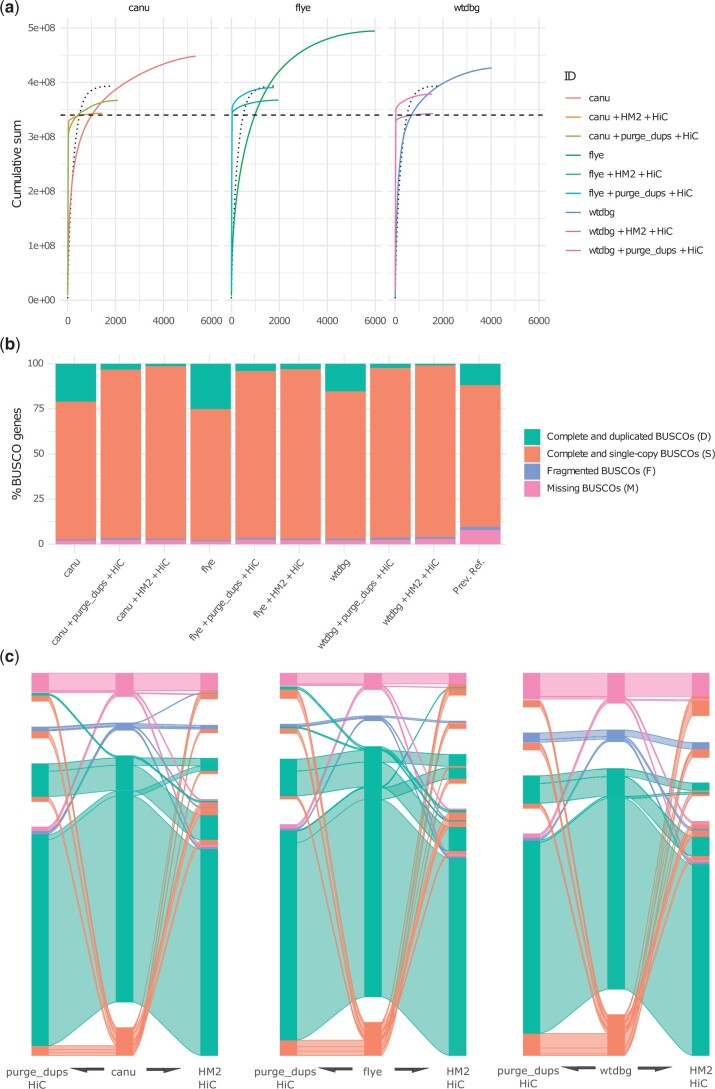
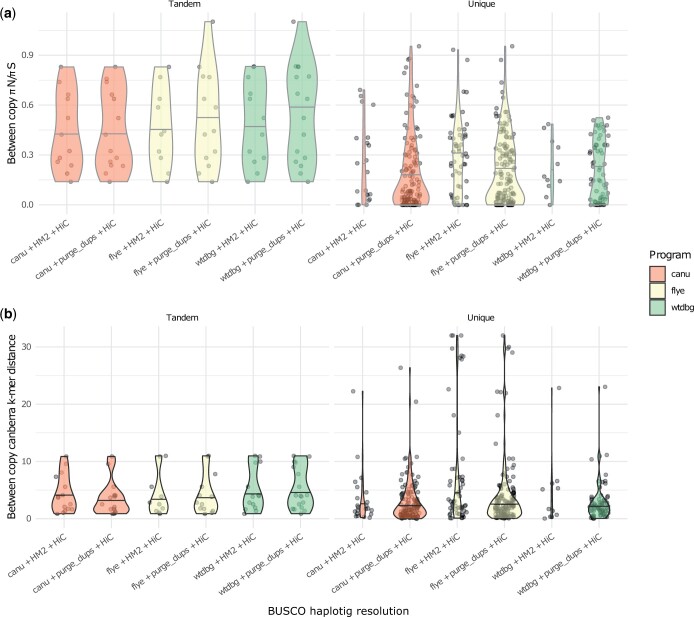
The assembly of divergent haplotypes using noisy long-read data presents a challenge to the reconstruction of haploid genome assemblies, due to overlapping distributions of technical sequencing error, intralocus genetic variation, and interlocus similarity within these data. Here, we present a comparative analysis of assembly algorithms representing overlap-layout-consensus, repeat graph, and de Bruijn graph methods. We examine how postprocessing strategies attempting to reduce redundant heterozygosity interact with the choice of initial assembly algorithm and ultimately produce a series of chromosome-level assemblies for an agricultural pest, the diamondback moth, Plutella xylostella (L.). We compare evaluation methods and show that BUSCO analyses may overestimate haplotig removal processing in long-read draft genomes, in comparison to a k-mer method. We discuss the trade-offs inherent in assembly algorithm and curation choices and suggest that "best practice" is research question dependent. We demonstrate a link between allelic divergence and allele-derived contig redundancy in final genome assemblies and document the patterns of coding and noncoding diversity between redundant sequences. We also document a link between an excess of nonsynonymous polymorphism and haplotigs that are unresolved by assembly or postassembly algorithms. Finally, we discuss how this phenomenon may have relevance for the usage of noisy long-read genome assemblies in comparative genomics.
Keywords: Plutella xylostella; assembly; haplotype; pool-seq.
© The Author(s) 2022. Published by Oxford University Press on behalf of Genetics Society of America.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous