

Subtype and cell type specific expression of lncRNAs provide insight into breast cancer
- PMID: 35982125
- PMCID: PMC9388662
- DOI: 10.1038/s42003-022-03559-7
Subtype and cell type specific expression of lncRNAs provide insight into breast cancer
Abstract
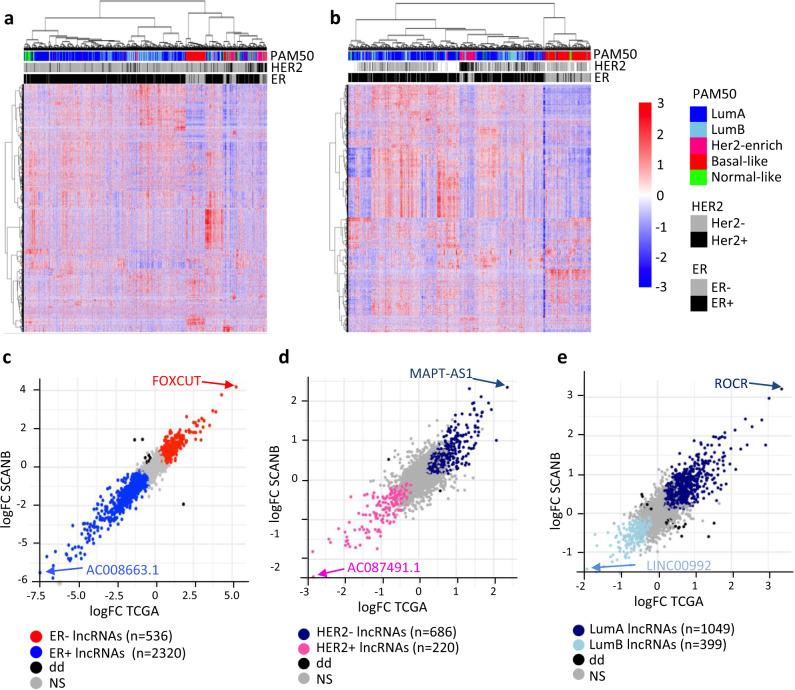
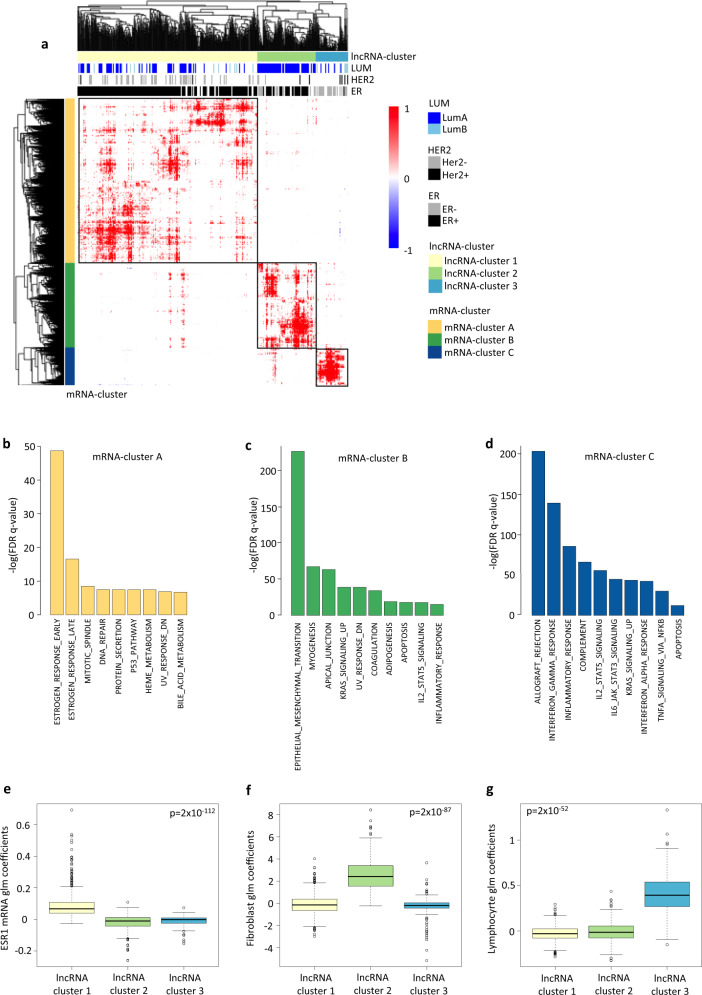
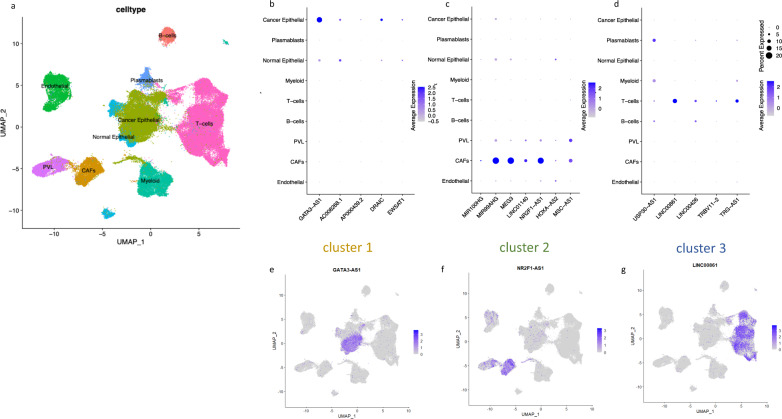
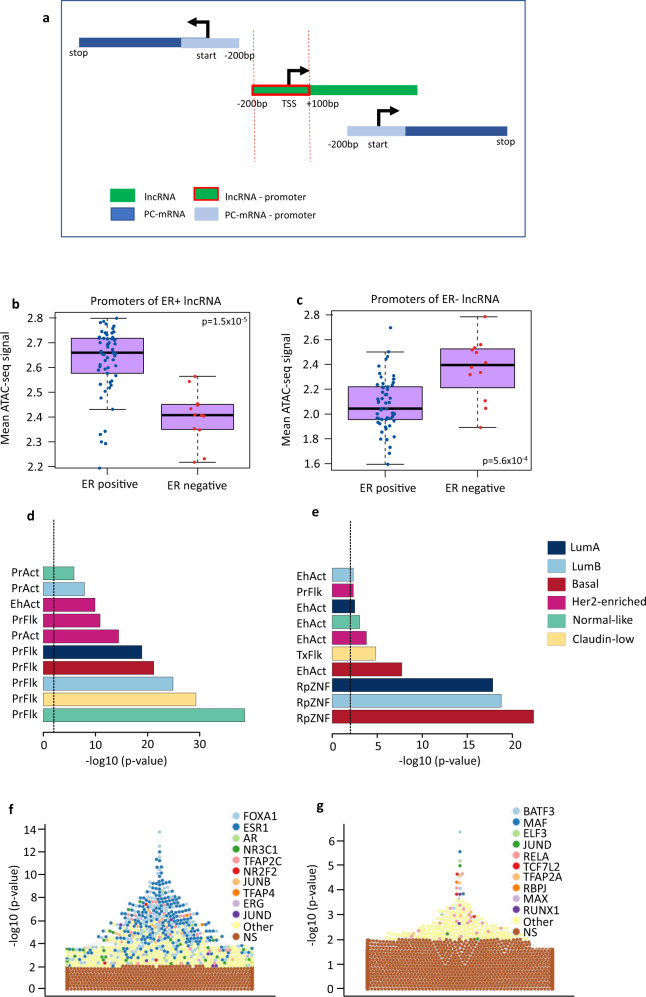
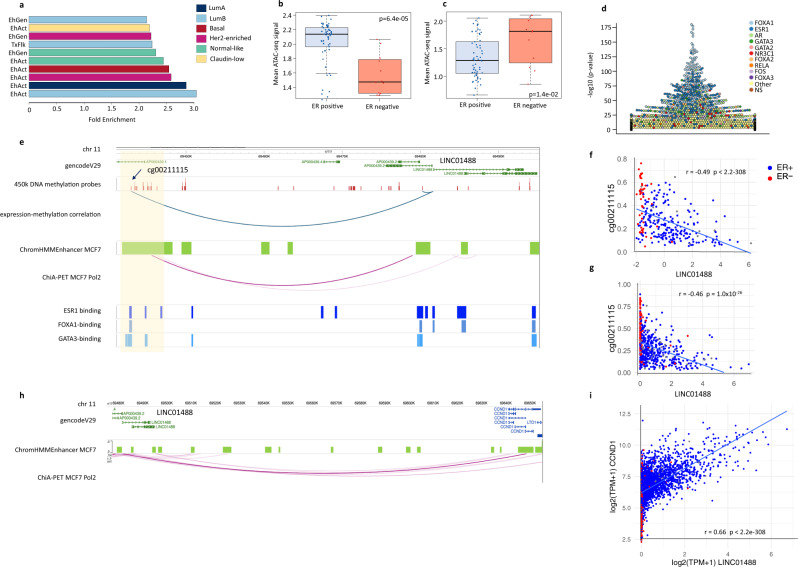
Long non-coding RNAs (lncRNAs) are involved in breast cancer pathogenesis through chromatin remodeling, transcriptional and post-transcriptional gene regulation. We report robust associations between lncRNA expression and breast cancer clinicopathological features in two population-based cohorts: SCAN-B and TCGA. Using co-expression analysis of lncRNAs with protein coding genes, we discovered three distinct clusters of lncRNAs. In silico cell type deconvolution coupled with single-cell RNA-seq analyses revealed that these three clusters were driven by cell type specific expression of lncRNAs. In one cluster lncRNAs were expressed by cancer cells and were mostly associated with the estrogen signaling pathways. In the two other clusters, lncRNAs were expressed either by immune cells or fibroblasts of the tumor microenvironment. To further investigate the cis-regulatory regions driving lncRNA expression in breast cancer, we identified subtype-specific transcription factor (TF) occupancy at lncRNA promoters. We also integrated lncRNA expression with DNA methylation data to identify long-range regulatory regions for lncRNA which were validated using ChiA-Pet-Pol2 loops. lncRNAs play an important role in shaping the gene regulatory landscape in breast cancer. We provide a detailed subtype and cell type-specific expression of lncRNA, which improves the understanding of underlying transcriptional regulation in breast cancer.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
