

Genetic control of RNA splicing and its distinct role in complex trait variation
- PMID: 35982161
- PMCID: PMC9470536
- DOI: 10.1038/s41588-022-01154-4
Genetic control of RNA splicing and its distinct role in complex trait variation
Abstract
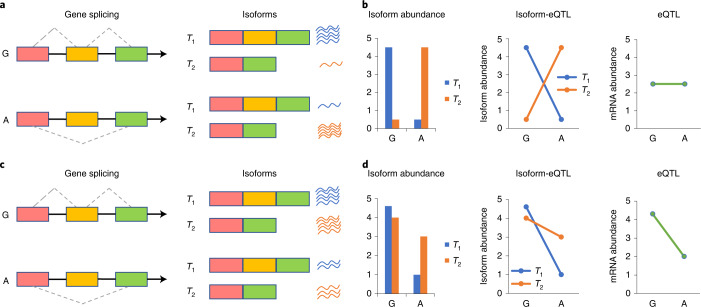
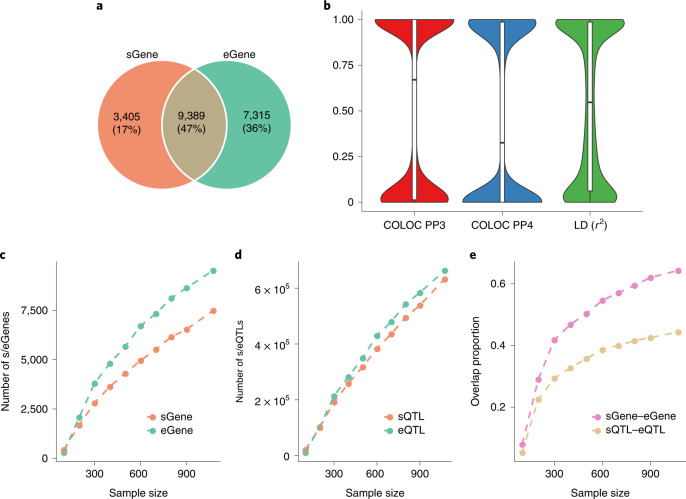
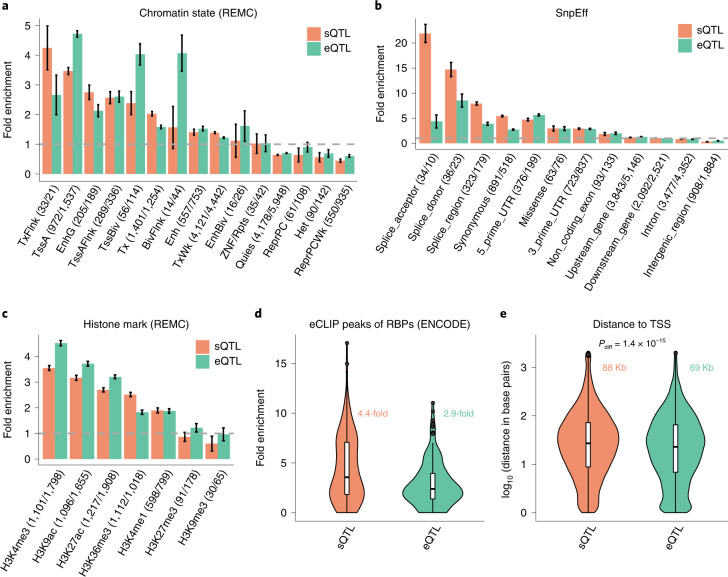
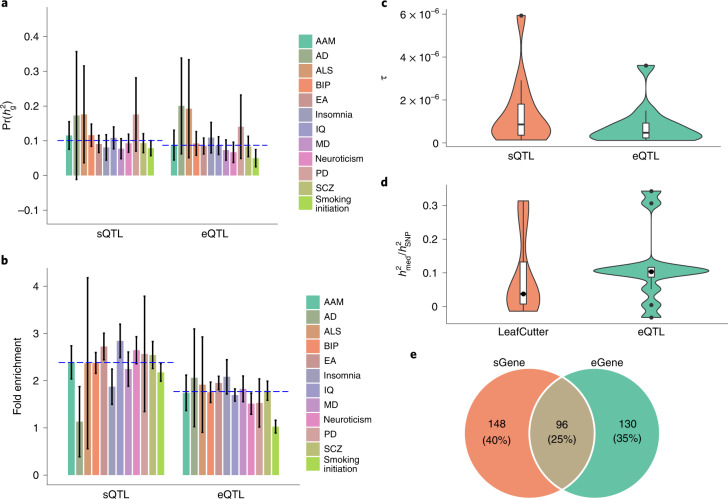
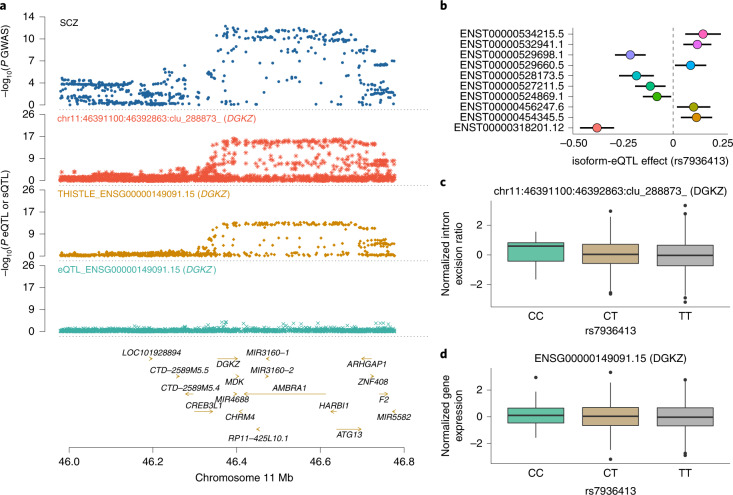
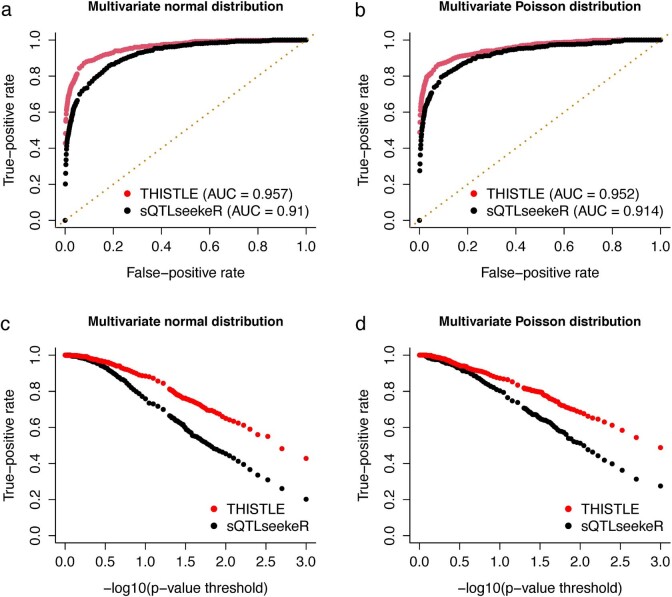
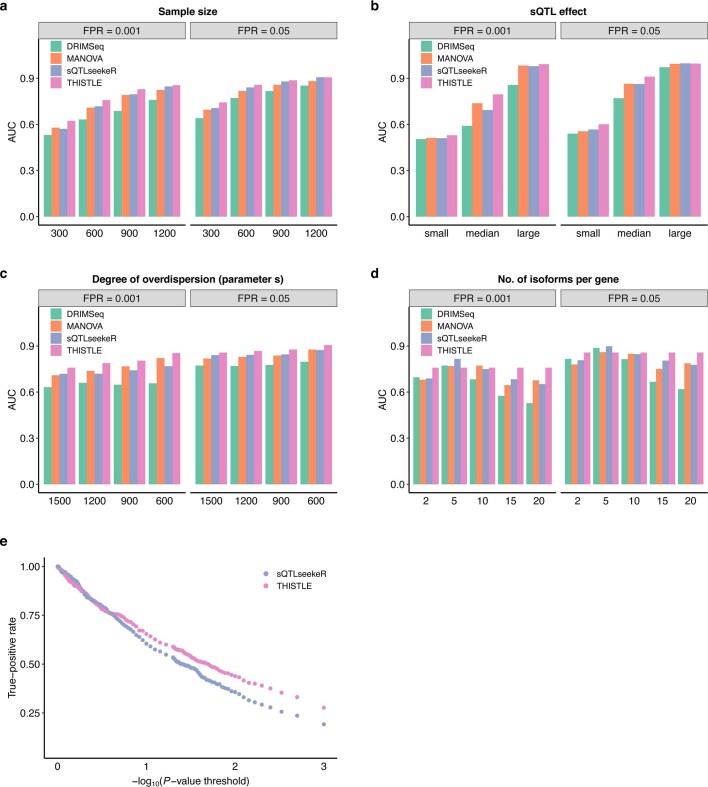
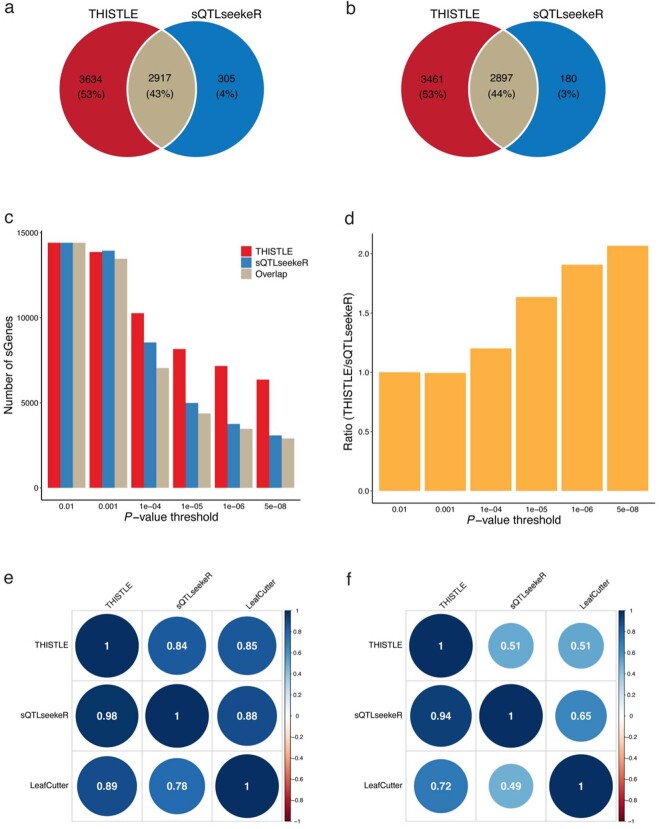
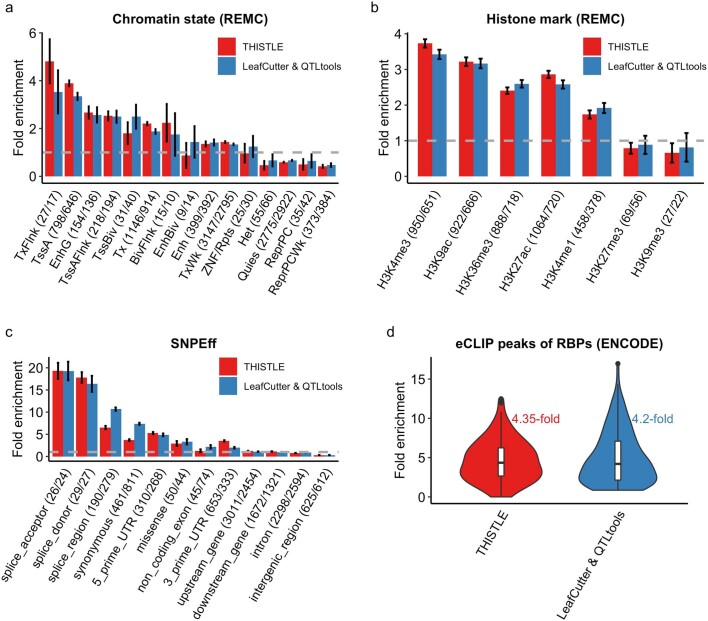
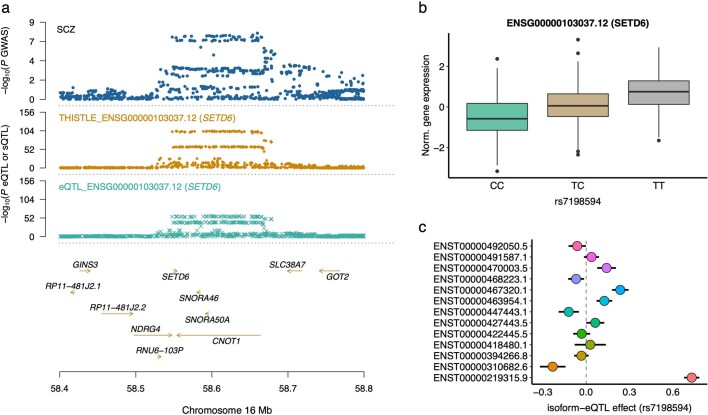
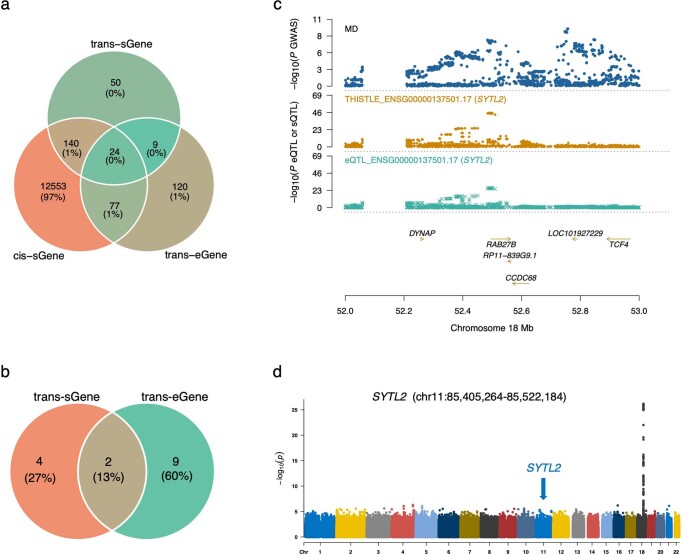
Most genetic variants identified from genome-wide association studies (GWAS) in humans are noncoding, indicating their role in gene regulation. Previous studies have shown considerable links of GWAS signals to expression quantitative trait loci (eQTLs) but the links to other genetic regulatory mechanisms, such as splicing QTLs (sQTLs), are underexplored. Here, we introduce an sQTL mapping method, testing for heterogeneity between isoform-eQTL effects (THISTLE), with improved power over competing methods. Applying THISTLE together with a complementary sQTL mapping strategy to brain transcriptomic (n = 2,865) and genotype data, we identified 12,794 genes with cis-sQTLs at P < 5 × 10-8, approximately 61% of which were distinct from eQTLs. Integrating the sQTL data into GWAS for 12 brain-related complex traits (including diseases), we identified 244 genes associated with the traits through cis-sQTLs, approximately 61% of which could not be discovered using the corresponding eQTL data. Our study demonstrates the distinct role of most sQTLs in the genetic regulation of transcription and complex trait variation.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
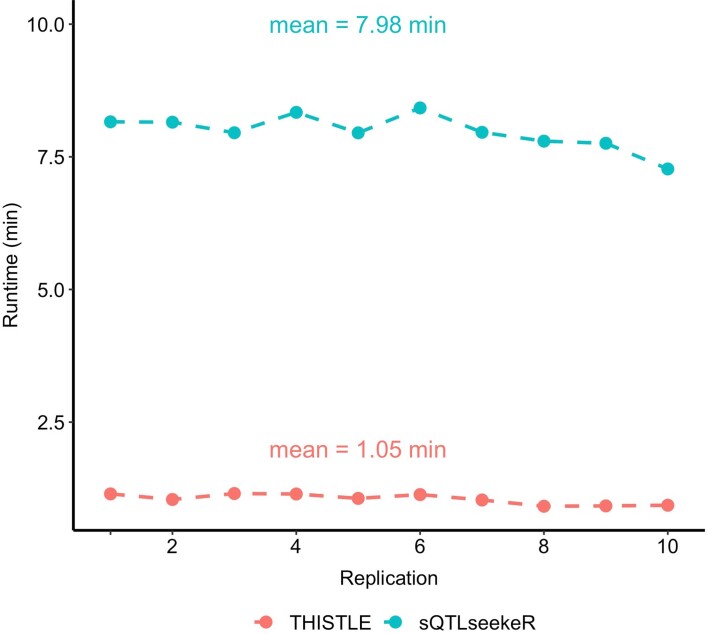
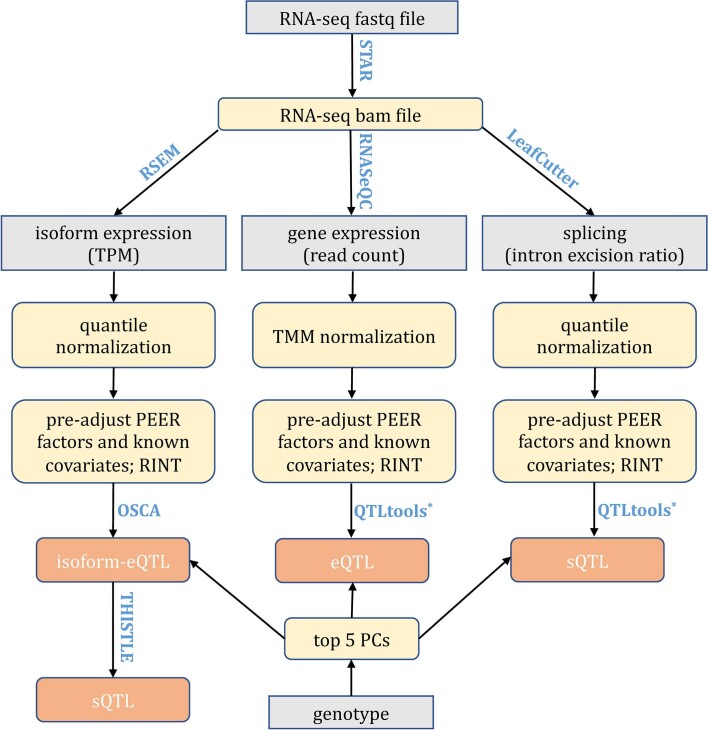
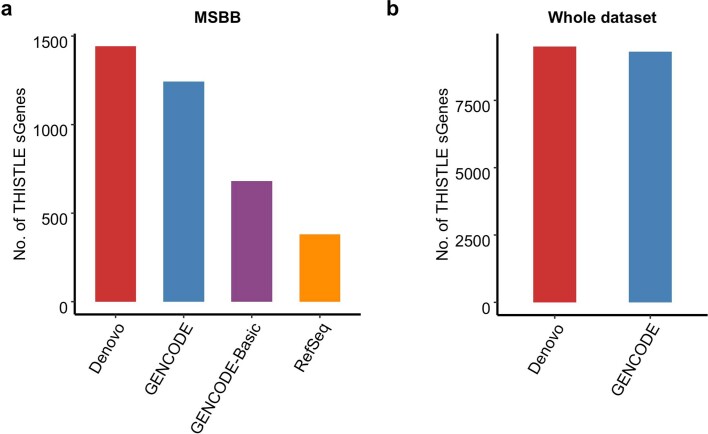
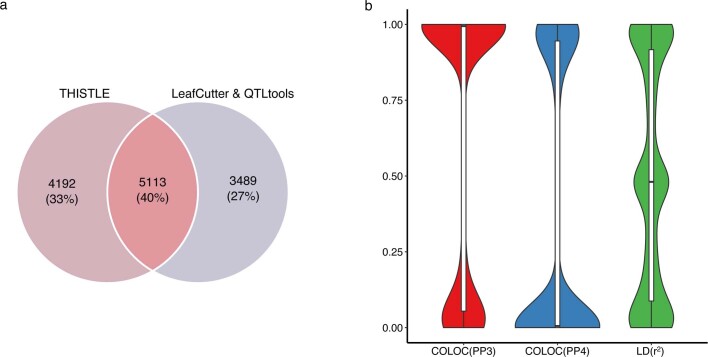
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
