

This is a preprint.
Within-host diversity of SARS-CoV-2 lineages and effect of vaccination
- PMID: 35982671
- PMCID: PMC9387541
- DOI: 10.21203/rs.3.rs-1927944/v1
Within-host diversity of SARS-CoV-2 lineages and effect of vaccination
Update in
-
Within-host genetic diversity of SARS-CoV-2 lineages in unvaccinated and vaccinated individuals.Nat Commun. 2023 Mar 31;14(1):1793. doi: 10.1038/s41467-023-37468-y. Nat Commun. 2023. PMID: 37002233 Free PMC article.
Abstract
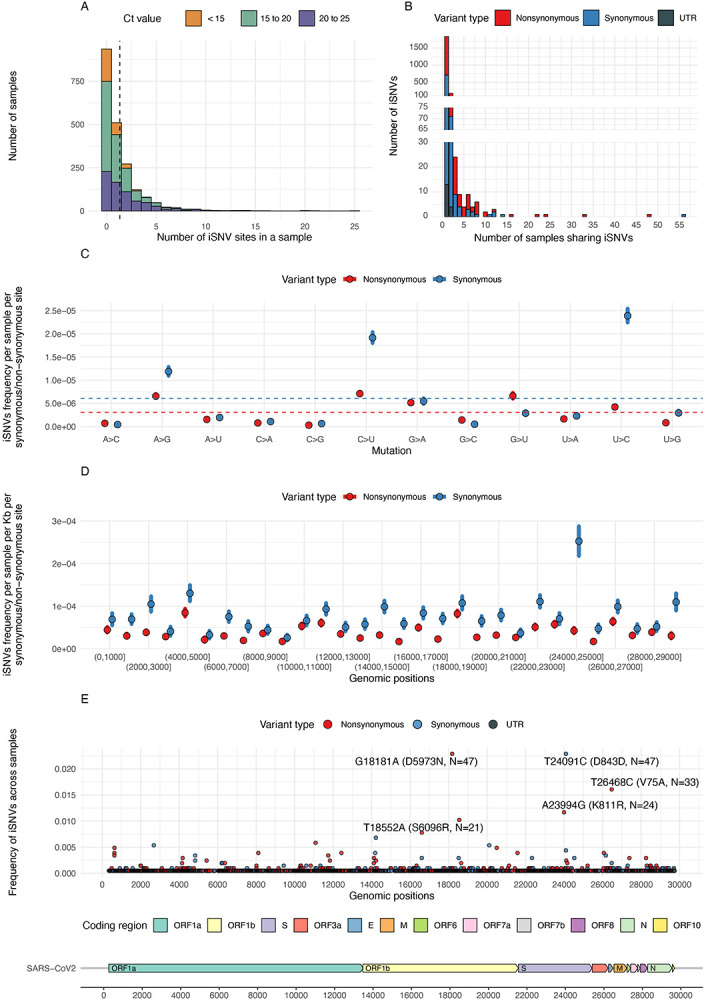
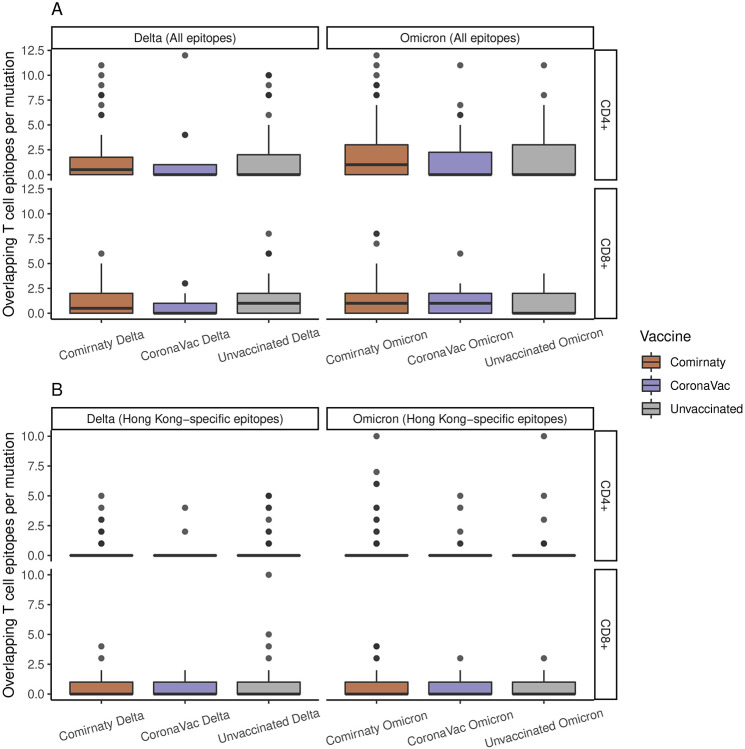
Viral and host factors can shape SARS-CoV-2 within-host viral diversity and virus evolution. However, little is known about lineage-specific and vaccination-specific mutations that occur within individuals. Here we analysed deep sequencing data from 2,146 SARS-CoV-2 samples with different viral lineages to describe the patterns of within-host diversity in different conditions, including vaccine-breakthrough infections. Variant of Concern (VOC) Alpha, Delta, and Omicron samples were found to have higher within-host nucleotide diversity while being under weaker purifying selection at full genome level compared to non-VOC SARS-CoV-2 viruses. Breakthrough Delta and Omicron infections in Comirnaty and CoronaVac vaccinated individuals appeared to have higher within-host purifying selection at the full-genome and/or Spike gene levels. Vaccine-induced antibody or T cell responses did not appear to have significant impact on within-host SARS-CoV-2 evolution. Our findings suggest that vaccination does not increase SARS-CoV-2 protein sequence space and may not facilitate emergence of more viral variants.
Figures



References
-
- Ritchie H., et al. Coronavirus pandemic (COVID-19). Our world in data (2020).
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
