

Discovery of small molecule ligands for the von Hippel-Lindau (VHL) E3 ligase and their use as inhibitors and PROTAC degraders
- PMID: 35983982
- PMCID: PMC9528729
- DOI: 10.1039/d2cs00387b
Discovery of small molecule ligands for the von Hippel-Lindau (VHL) E3 ligase and their use as inhibitors and PROTAC degraders
Abstract
The von Hippel-Lindau (VHL) Cullin RING E3 ligase is an essential enzyme in the ubiquitin-proteasome system that recruits substrates such as the hypoxia inducible factor for ubiquitination and subsequent proteasomal degradation. The ubiquitin-proteasome pathway can be hijacked toward non-native neo-substrate proteins using proteolysis targeting chimeras (PROTACs), bifunctional molecules designed to simultaneously bind to an E3 ligase and a target protein to induce target ubiquitination and degradation. The availability of high-quality small-molecule ligands with good binding affinity for E3 ligases is fundamental for PROTAC development. Lack of good E3 ligase ligands as starting points to develop PROTAC degraders was initially a stumbling block to the development of the field. Herein, the journey towards the design of small-molecule ligands binding to VHL is presented. We cover the structure-based design of VHL ligands, their application as inhibitors in their own right, and their implementation into rationally designed, potent PROTAC degraders of various target proteins. We highlight the key findings and learnings that have provided strong foundations for the remarkable development of targeted protein degradation, and that offer a blueprint for designing new ligands for E3 ligases beyond VHL.
Conflict of interest statement
The Ciulli laboratory receives or has received sponsored research support from Almirall, Amgen, Amphista Therapeutics, Boehringer Ingelheim, Eisai, Merck KGaA, Nurix Therapeutics, Ono Pharmaceutical and Tocris-BioTechne. A. C. is a scientific founder, advisor, and shareholder of Amphista Therapeutics, a company that is developing targeted protein degradation therapeutic platforms. C. D. reports no competing interest.
Figures









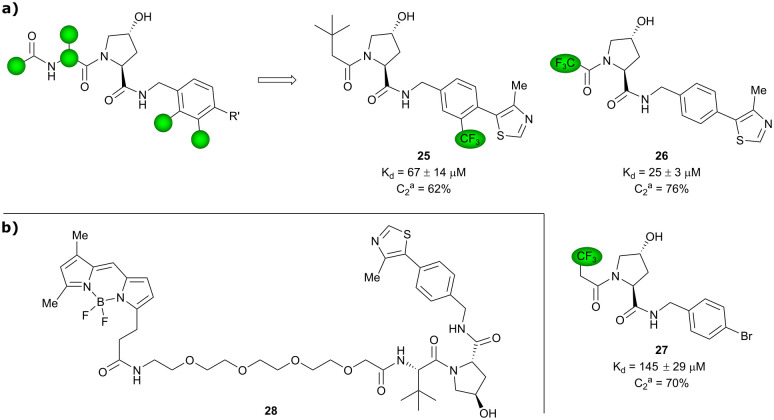
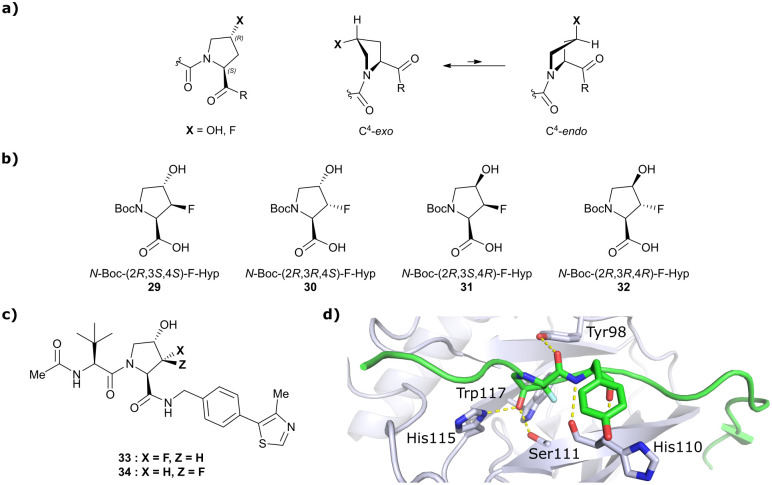
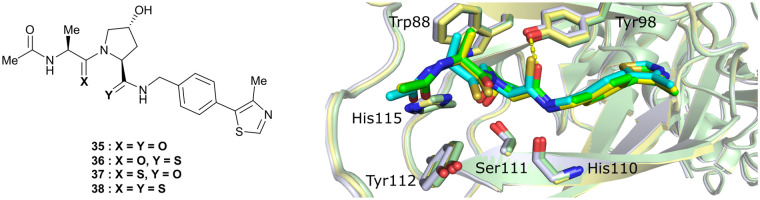


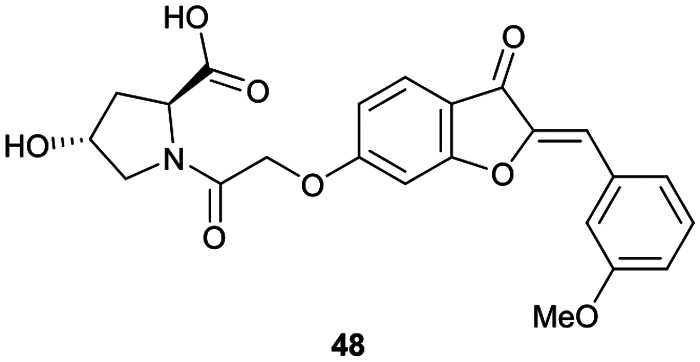









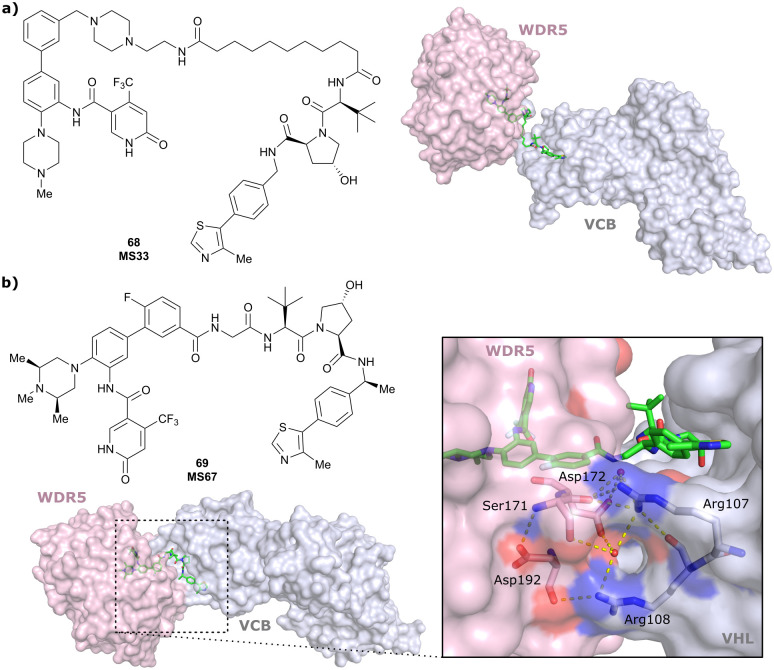



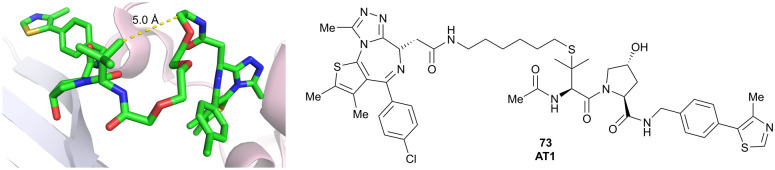
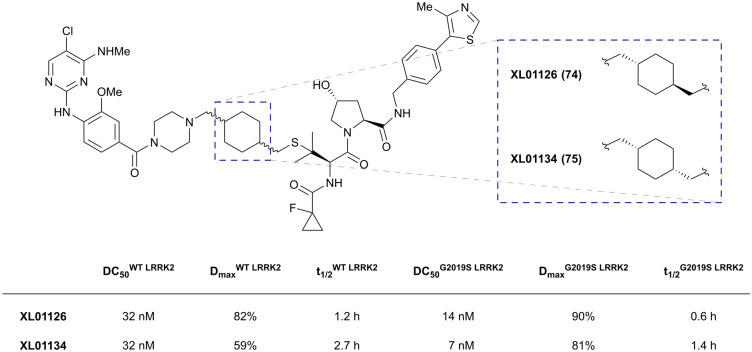





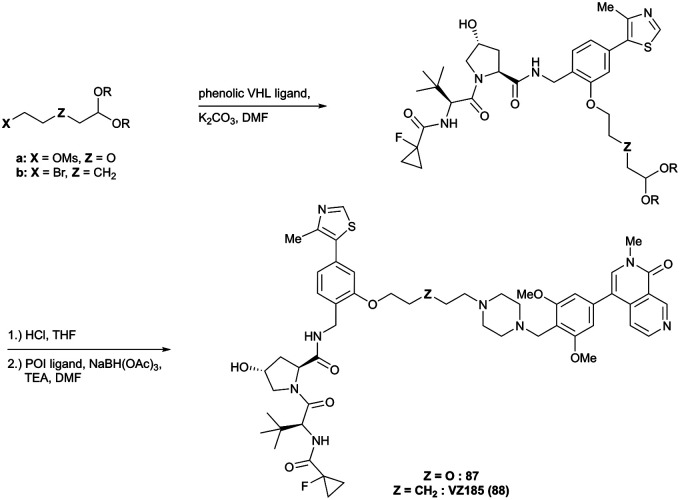
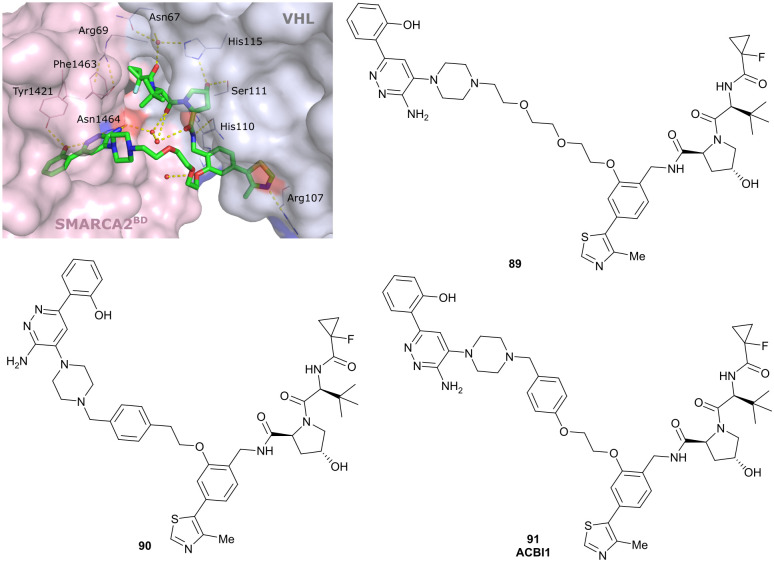


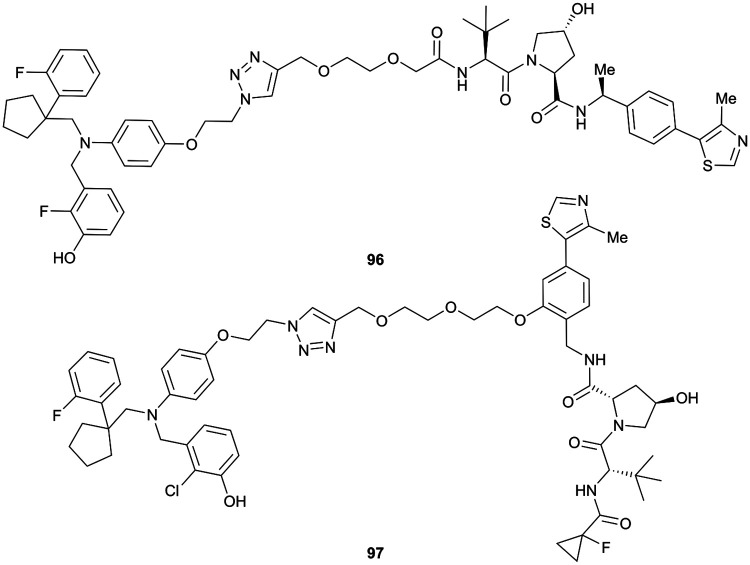






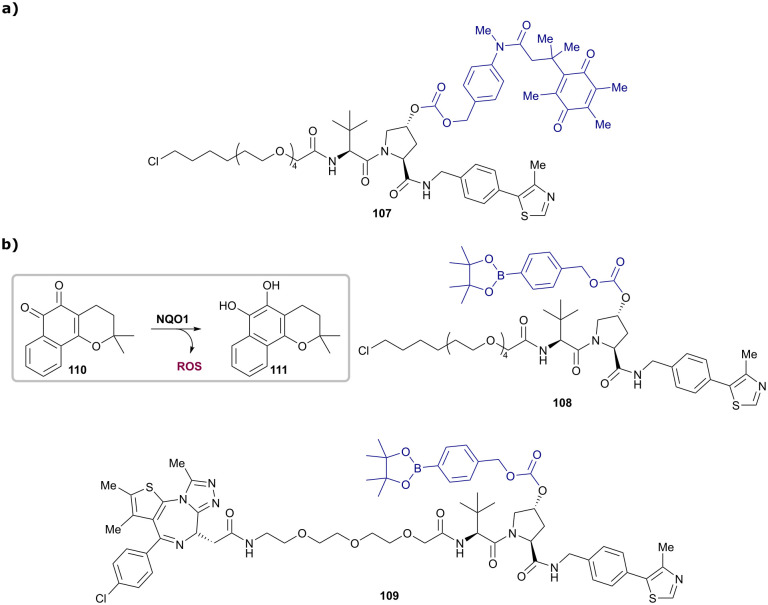
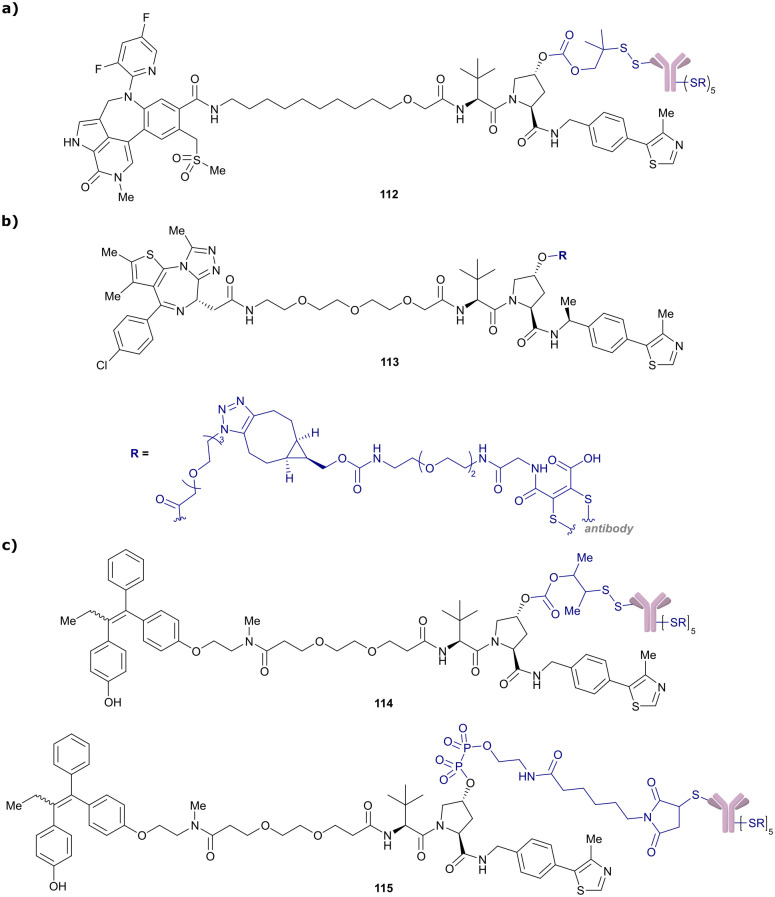




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources