

TreeKnit: Inferring ancestral reassortment graphs of influenza viruses
- PMID: 35984845
- PMCID: PMC9447925
- DOI: 10.1371/journal.pcbi.1010394
TreeKnit: Inferring ancestral reassortment graphs of influenza viruses
Abstract
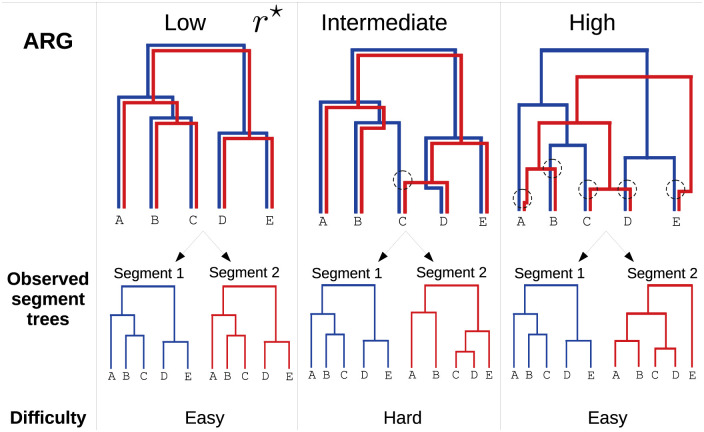
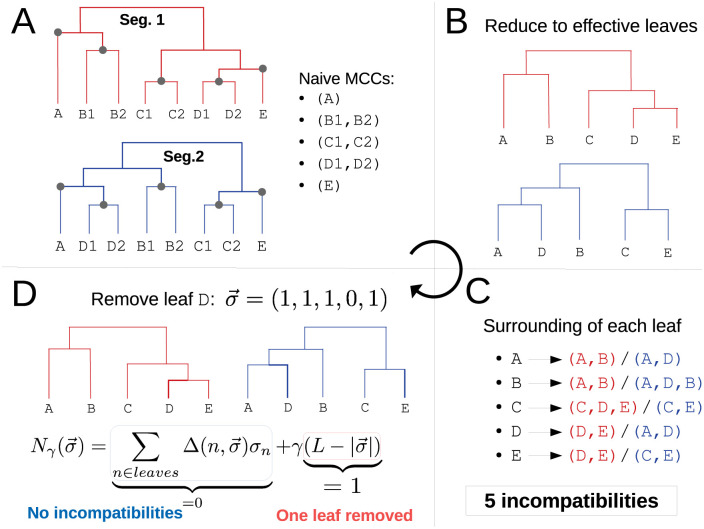
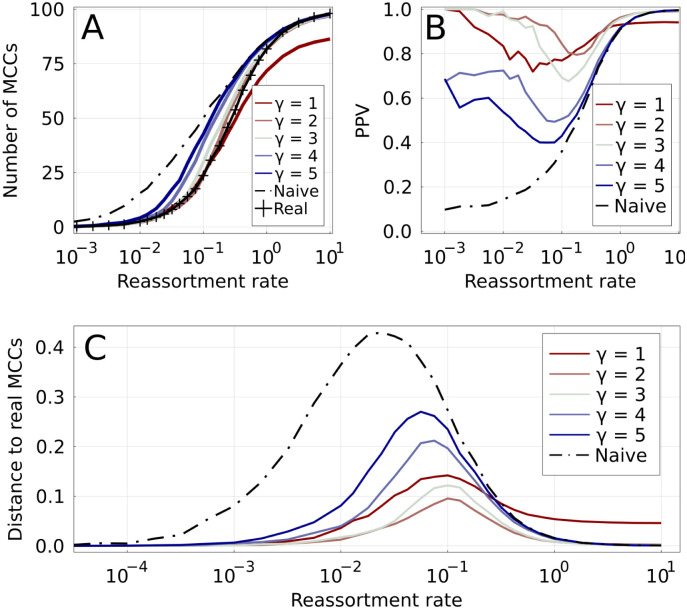
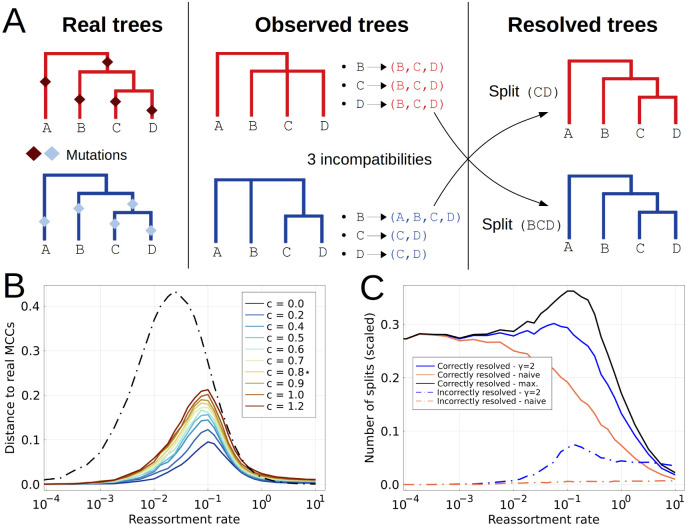
When two influenza viruses co-infect the same cell, they can exchange genome segments in a process known as reassortment. Reassortment is an important source of genetic diversity and is known to have been involved in the emergence of most pandemic influenza strains. However, because of the difficulty in identifying reassortment events from viral sequence data, little is known about their role in the evolution of the seasonal influenza viruses. Here we introduce TreeKnit, a method that infers ancestral reassortment graphs (ARG) from two segment trees. It is based on topological differences between trees, and proceeds in a greedy fashion by finding regions that are compatible in the two trees. Using simulated genealogies with reassortments, we show that TreeKnit performs well in a wide range of settings and that it is as accurate as a more principled bayesian method, while being orders of magnitude faster. Finally, we show that it is possible to use the inferred ARG to better resolve segment trees and to construct more informative visualizations of reassortments.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
