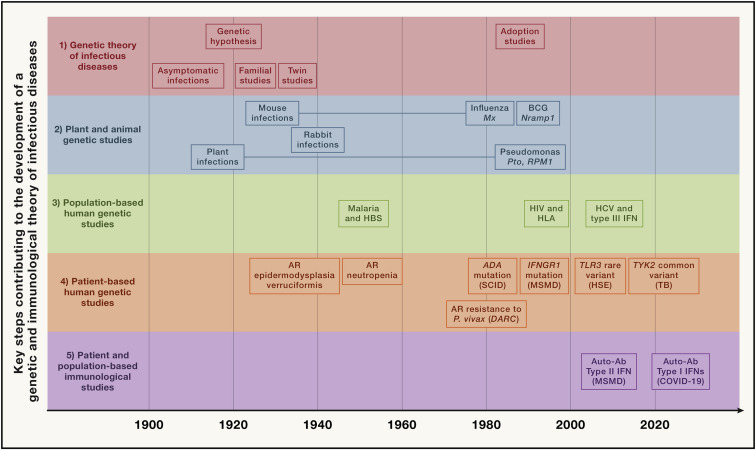
Figure 1
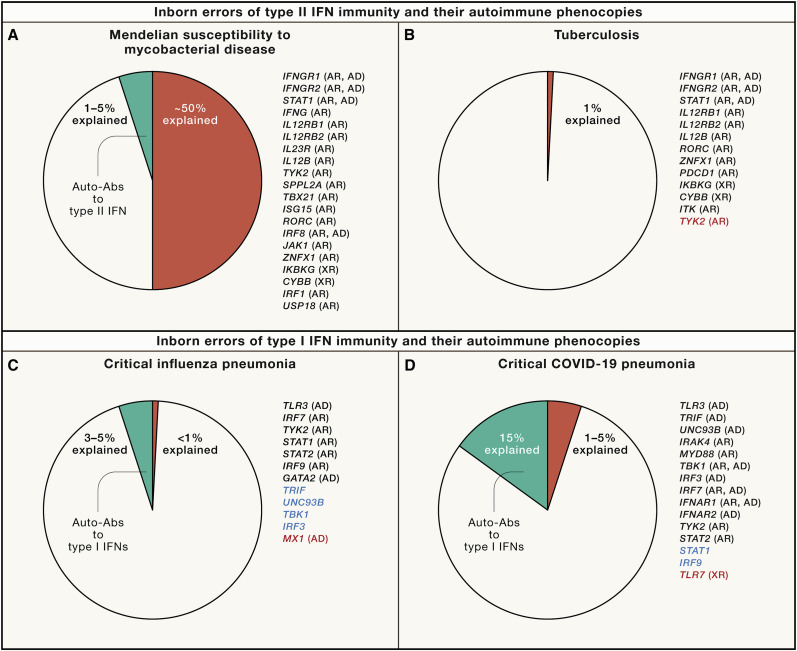
Timeline showing some of the key steps that have progressively contributed to the development of a genetic and immunological theory of infectious diseases, grouped into five main parallel fields, from top to bottom (1) Germ and genetic theories of infectious diseases (red). Following the establishment of the germ theory of disease between 1865 and 1870 (Louis Pasteur) (Pasteur, 1922–1939) and its radical version with Koch’s postulates in 1882 (Robert Koch) (Koch, 1882), it became widely accepted that life-threatening fevers in animals and humans were infectious. Chief among Koch’s postulates was the notion that a pathogen must be found in all patients with a given disease and not in healthy individuals. Once established, this postulate made it difficult to appreciate and understand the importance of the evidence of both latent (non-replicating, dormant germs in tissues of asymptomatic individuals; Clemens von Pirquet and followers) (von Pirquet, 1909) and unapparent (replicating germs in tissues or bloodstream of asymptomatic individuals; Charles Nicolle) (Nicolle, 1933) silent infections. Human geneticists proposed a genetic solution to the problem of asymptomatic infection. Karl Pearson (Pearson, 1912) and other population geneticists and biometricians, in parallel with Archibald Garrod (Garrod, 1931) and other clinical and biochemical geneticists, proposed that the germline genetic background of the host influences or determines susceptibility or resistance to any given microbe. J.B.S. Haldane proposed that infectious diseases had played a major role in natural selection (Haldane, 1949). Various epidemiological and familial approaches were conducted in the first half of the 20th century, supporting this hypothesis. The most remarkable and convincing investigations were probably twin studies comparing monozygotic and dizygotic twins for concordance for a particular infectious phenotype, in particular for tuberculosis (TB) (Puffer, 1944). Adoption studies, which compare the cause of death of patients with that of their biological and foster parents and are equally powerful, were conducted later (Sørensen et al., 1988). (2) Plant and animal genetic studies (blue). Plant biologists and geneticists also reported early in the 20th century that resistance or susceptibility to infection can be genetically determined (Biffen, 1905; Flor, 1942). From the 1920s onward, other researchers approached the question of the host genetics of infectious diseases from the angle of animal models, including mice and rabbits in particular. Their results led to a similar conclusion, as some strains were vulnerable to the infections tested, whereas others were not (Lurie, 1941; Webster, 1939). These were experimental, as opposed to natural, infections. The experiments were powerful. By the late 1930s, the stage was set for major progress and a transition to molecular genetics, building on multiple independent lines of thought and investigation. Molecular discoveries began in mice in 1986, with the identification, by molecular complementation, of the first monogenic infection susceptibility gene, Mx, mutations of which underlie influenza virus infections (Staeheli et al., 1986), followed by the discoveries, by positional cloning, of the Bcg (Nramp1) (Skamene et al., 1982; Vidal et al., 1993), Cmv (Ly49h) (Brown et al., 2001), and Lps (TLR4) (Poltorak et al., 1998) loci. In parallel, superb studies unraveled the monogenic basis of various infections in various plant species (Dangl and Jones, 2001; Jones et al., 2016), including, in particular, the role of the Pto and RPM1 genes in resistance to Pseudomonas in tomato and Arabidopsis thaliana, respectively (Grant et al., 1995; Martin et al., 1993). (3) Population-based human genetic studies (green). At the population level, the field of human genetics of infectious diseases began with the discovery, by Allison in 1954, that the sickle cell trait (HbS) provides significant protection against severe forms of P. falciparum malaria (Allison, 1954). Classical genetic epidemiology studies, particularly segregation analyses followed by genome-wide linkage studies, identified major loci for common infectious conditions, such as schistosomiasis and leprosy (Abel and Demenais, 1988; Alcaïs et al., 2007; Marquet et al., 1996; Mira et al., 2004). Very few candidate gene association studies were successful, and only a handful reported odds ratios (ORs) for developing the disease >2 that were replicated in independent populations; among the most remarkable is the association of some HLA class I alleles with AIDS progression, with hazard ratios for protection of about 0.3 (Kaslow et al., 1996). Finally, genome-wide association studies (GWASs) met with variable success, depending on the infectious disease, with many loci identified in leprosy (Zhang et al., 2009, 2011) with ORs below 2, and, more recently, malaria (Malaria Genomic Epidemiology, 2019), with ORs for protection above 0.5, except for the HBB locus. The most remarkable achievement of GWAS in infectious diseases was probably the identification, toward 2010, of IL28B variants (IL28B encodes the type III IFN-λ3) strongly associated with the clearance of hepatitis C virus (Ge et al., 2009) (OR for clearance ∼6). More recently, GWAS in COVID-19 have identified several common variants associated with severe pneumonia, the most significant being located on chromosome 3 with an OR ∼2 (COVID-19 Host Genetics Initiative, 2021; Kousathanas et al., 2022; Pairo-Castineira et al., 2021). (4) Patient-based human genetic studies (orange). In the early 1950s, pediatricians and clinical geneticists described the first inborn errors of immunity (IEIs)—then referred to as primary immunodeficiencies (PIDs)—as rare, Mendelian, early-onset conditions underlying both multiple, recurrent, and opportunistic infections, and overt, or at least detectable, immunological abnormalities. The blueprint for conventional IEI is widely agreed to be the description of Bruton’s X-linked recessive (XR) agammaglobulinemia in 1952 (Bruton, 1952). Severe congenital neutropenia had, however, been described earlier, in 1950, in children with severe staphylococcal and other bacterial infections and congenital neutropenia, two phenotypes that co-segregated as an autosomal recessive (AR) trait (Kostmann, 1950, 1956). The first IEI conferring predisposition to a single infectious agent was, however, reported even earlier, in 1946, when Wilhelm Lutz described epidermodysplasia verruciformis (EV) as an AR predisposition to skin-tropic viruses (Lutz, 1946) identified in 1978 by Gérard Orth to be weakly virulent human papillomaviruses (HPVs) (Orth et al., 1978). Immunologists did not consider EV to be an IEI, because of the lack of a detectable leukocyte abnormality, until the discovery of EV-causing genes from 2002 onward led to the gradual recognition that keratinocytes contribute to host defense (de Jong et al., 2018; Ramoz et al., 2002). Defects of the membrane attack complex of complement (underlying infections with Neisseria), and X-linked lymphoproliferation (XLP, Epstein-Barr virus) were described later, from different angles, as XLP, like EV, was described as a Mendelian and unexplained (idiopathic) infection (Purtilo et al., 1974, 1977), whereas complement defects were found serendipitously in sporadic cases of Neisseria disease (Lim et al., 1976; Petersen et al., 1976). Mendelian resistance to infectious agents was first detected in the 1970s, with the Duffy antigen receptor for chemokines (DARCs) and P. vivax malaria (Miller et al., 1976), the genetic basis of which was elucidated in 1995 (Tournamille et al., 1995). It was followed, in the mid-1990s, by the discovery of the link between C-C chemokine receptor 5 (CCR5) variants and resistance to human immunodeficiency virus-1 (HIV-1) (Dean et al., 1996; Liu et al., 1996; Samson et al., 1996). The first mutated gene underlying a conventional IEI, ADA, was reported in patients with severe combined immunodeficiency in 1985 (Bonthron et al., 1985). The first mutated gene underlying an isolated infection, IFNGR1, was identified in patients with MSMD in 1996 (Jouanguy et al., 1996; Newport et al., 1996). Since 2007, some rare and sporadic infectious diseases (e.g., HSE) have been shown to be caused by rare monogenic defects, with incomplete penetrance (Casanova and Abel, 2021a; Zhang et al., 2007). Since 2001, rare monogenic defects have also been shown to underlie some common infectious diseases in rare patients (e.g., TB in 2001 [Altare et al., 2001] and COVID-19 in 2020 [Zhang et al., 2020]). Since 2018, monogenic causes of infection have been found in a greater proportion of patients with TB (due to homozygosity for a common TYK2 allele) (Boisson-Dupuis et al., 2018; Kerner et al., 2019, 2021) or COVID-19 (due to hemizygosity for various rare TLR7 alleles) (Asano et al., 2021; Casanova and Abel, 2021a). (5) Patient- and population-based immunological studies (magenta). Auto-Abs against four cytokines (type I and II IFNs, IL-6, and IL-17A/F) have been shown to underlie infectious phenocopies of the corresponding inborn errors of cytokines or their response pathways (Ku et al., 2020; Puel et al., 2022). The best characterized are probably auto-Abs neutralizing type II IFN, which underlie a phenocopy of MSMD and, more rarely, TB (Puel et al., 2022; Shih et al., 2021). Auto-Abs neutralizing type I IFNs, first described in the early 1980s, were long thought to be clinically silent, except for a 77-year-old woman with disseminated zoster studied by Ion Gresser (Pozzetto et al., 1984). They were found in almost all patients with autoimmune polyendocrine syndrome type 1 (APS-1) (Husebye et al., 2018; Meager et al., 2006). Since 2020, they have been shown to be strong determinants of at least 15% of cases of life-threatening COVID-19 pneumonia (Bastard et al., 2020, 2021a; Zhang et al., 2022a) and a third of adverse reactions to the live-attenuated yellow fever vaccine (Bastard et al., 2021c). More recently, they have also been shown to account for ∼20% of hypoxemic breakthrough COVID-19 cases in fully vaccinated subjects (Bastard et al., 2022b), and ∼5% of cases of severe influenza pneumonia in patients younger than 70 years (Zhang et al., 2022).