

A druggable addiction to de novo pyrimidine biosynthesis in diffuse midline glioma
- PMID: 35985342
- PMCID: PMC9575661
- DOI: 10.1016/j.ccell.2022.07.012
A druggable addiction to de novo pyrimidine biosynthesis in diffuse midline glioma
Abstract
Diffuse midline glioma (DMG) is a uniformly fatal pediatric cancer driven by oncohistones that do not readily lend themselves to drug development. To identify druggable targets for DMG, we conducted a genome-wide CRISPR screen that reveals a DMG selective dependency on the de novo pathway for pyrimidine biosynthesis. This metabolic vulnerability reflects an elevated rate of uridine/uracil degradation that depletes DMG cells of substrates for the alternate salvage pyrimidine biosynthesis pathway. A clinical stage inhibitor of DHODH (rate-limiting enzyme in the de novo pathway) diminishes uridine-5'-phosphate (UMP) pools, generates DNA damage, and induces apoptosis through suppression of replication forks-an "on-target" effect, as shown by uridine rescue. Matrix-assisted laser desorption/ionization (MALDI) mass spectroscopy imaging demonstrates that this DHODH inhibitor (BAY2402234) accumulates in the brain at therapeutically relevant concentrations, suppresses de novo pyrimidine biosynthesis in vivo, and prolongs survival of mice bearing intracranial DMG xenografts, highlighting BAY2402234 as a promising therapy against DMGs.
Keywords: ATR; BAY2402234; DHODH; DPYD; de novo pyrimidine synthesis; diffuse intrinsic pontine glioma; diffuse midline glioma; elimusertib; pyrimidine degradation; replication stress.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests N.Y.R.A. is key opinion leader for Bruker Daltonics, scientific advisor to Invicro, and receives support from Thermo Finnegan and EMD Serono. S.K.M. has served as a paid advisor to Agios Pharmaceuticals. The other authors declare no competing interests.
Figures

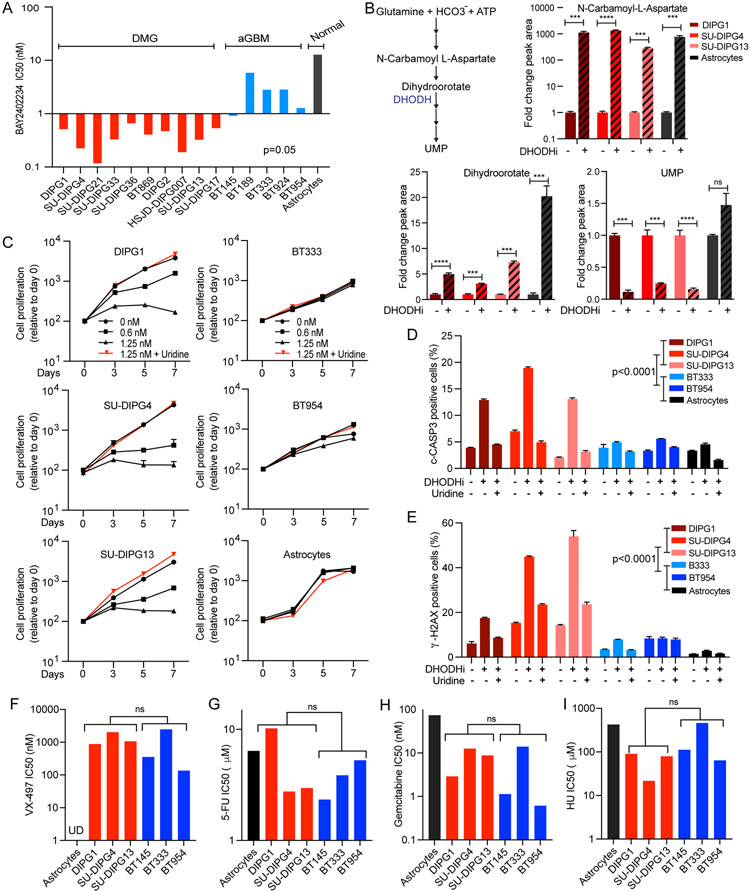

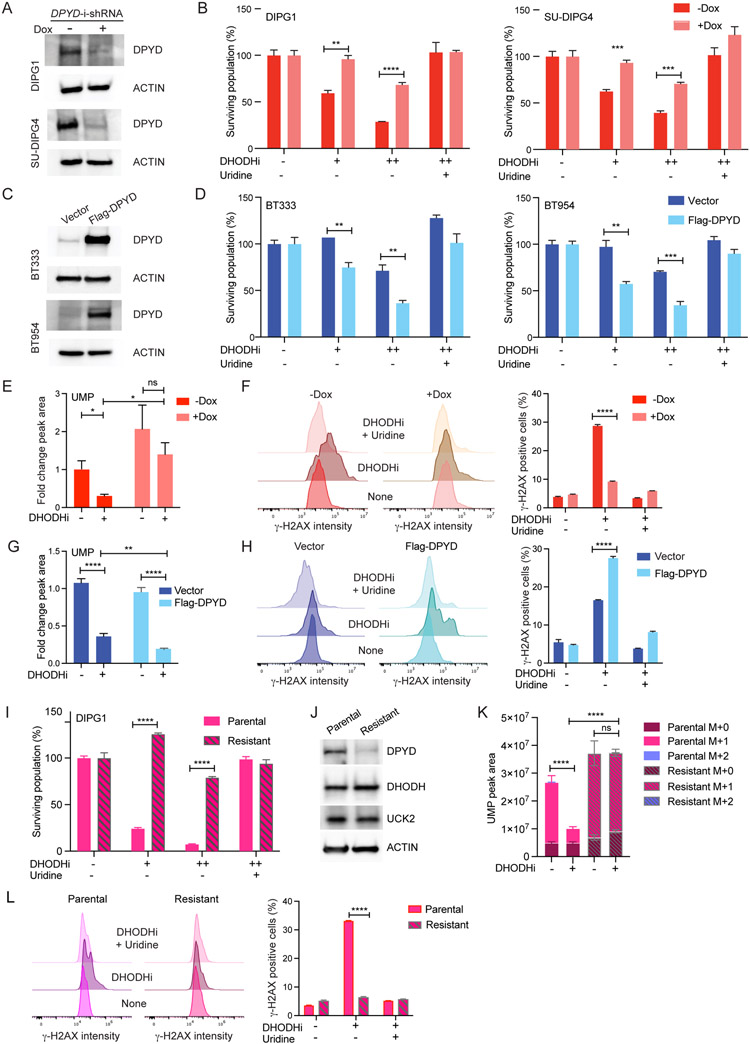
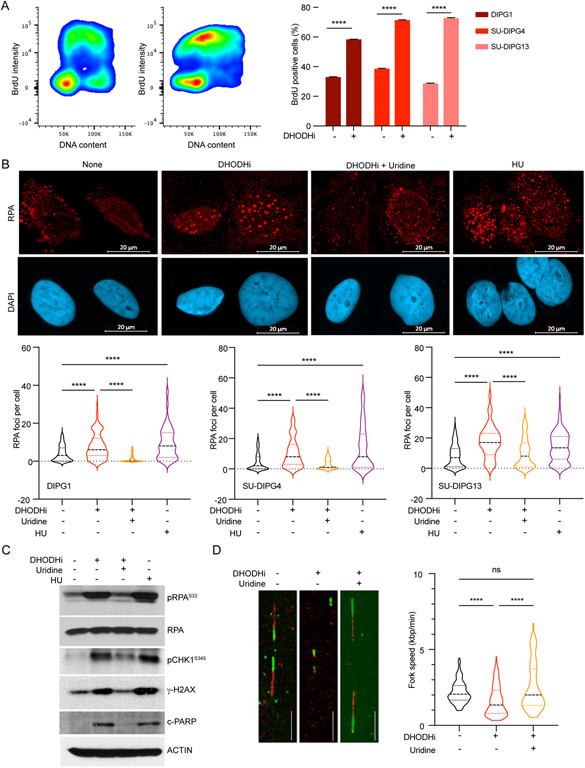
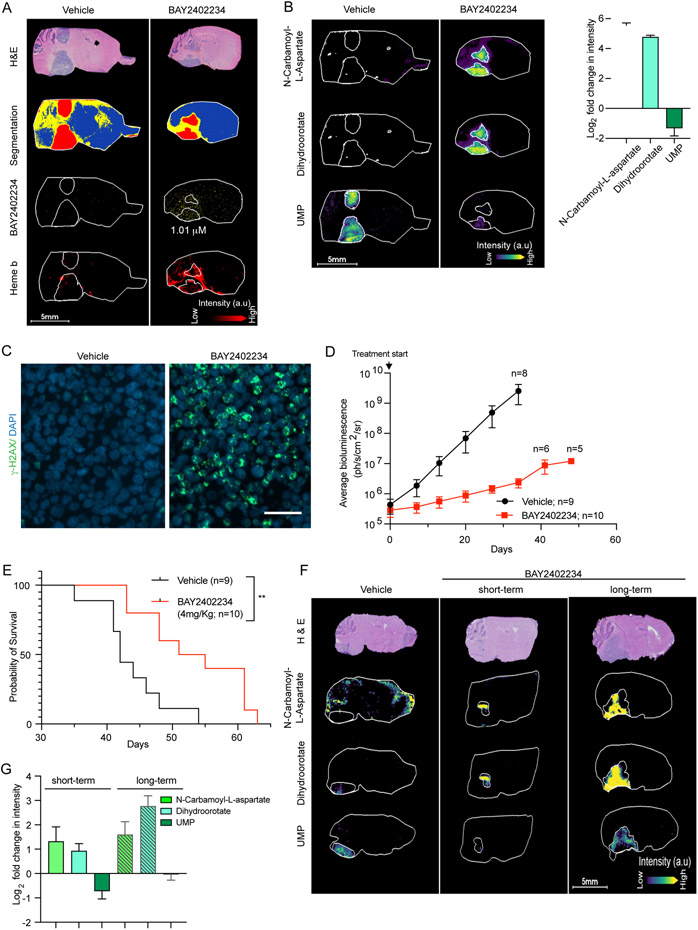

Comment in
-
Gliomas lean on pyrimidines.Nat Rev Cancer. 2022 Nov;22(11):606-607. doi: 10.1038/s41568-022-00515-9. Nat Rev Cancer. 2022. PMID: 36131135 No abstract available.
References
-
- Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, et al. (2013). Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24, 660–672. - PubMed
-
- Braunstein S, Raleigh D, Bindra R, Mueller S, and Haas-Kogan D (2017). Pediatric high-grade glioma: current molecular landscape and therapeutic approaches. J Neurooncol 134, 541–549. - PubMed
-
- Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E, Bartels U, et al. (2014). Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 46, 451–456. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
