

Genetic and modifiable risk factors combine multiplicatively in common disease
- PMID: 35987817
- PMCID: PMC9898372
- DOI: 10.1007/s00392-022-02081-4
Genetic and modifiable risk factors combine multiplicatively in common disease
Abstract
Background: The joint contribution of genetic and environmental exposures to noncommunicable diseases is not well characterized.
Objectives: We modeled the cumulative effects of common risk alleles and their prevalence variations with classical risk factors.
Methods: We analyzed mathematically and statistically numbers and effect sizes of established risk alleles for coronary artery disease (CAD) and other conditions.
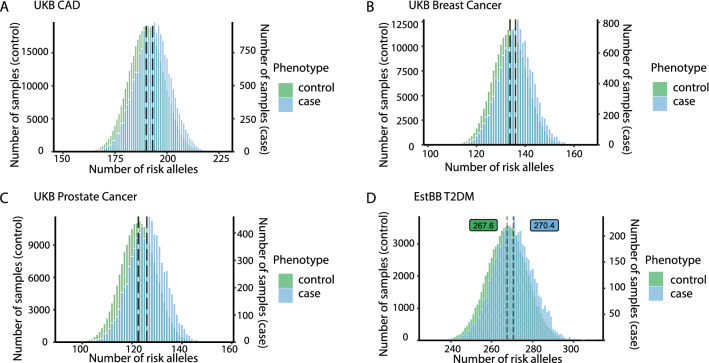
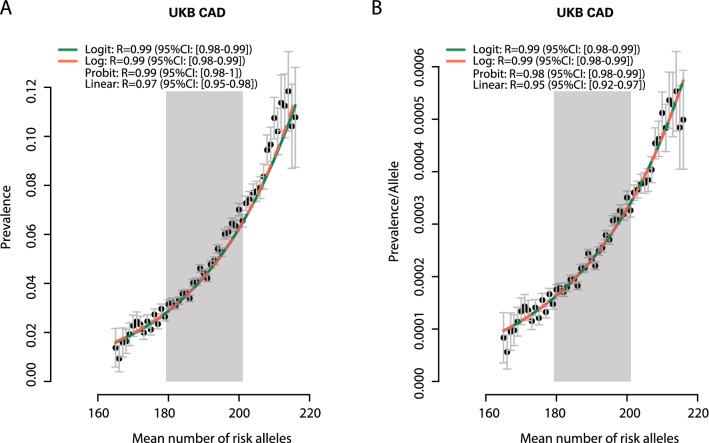
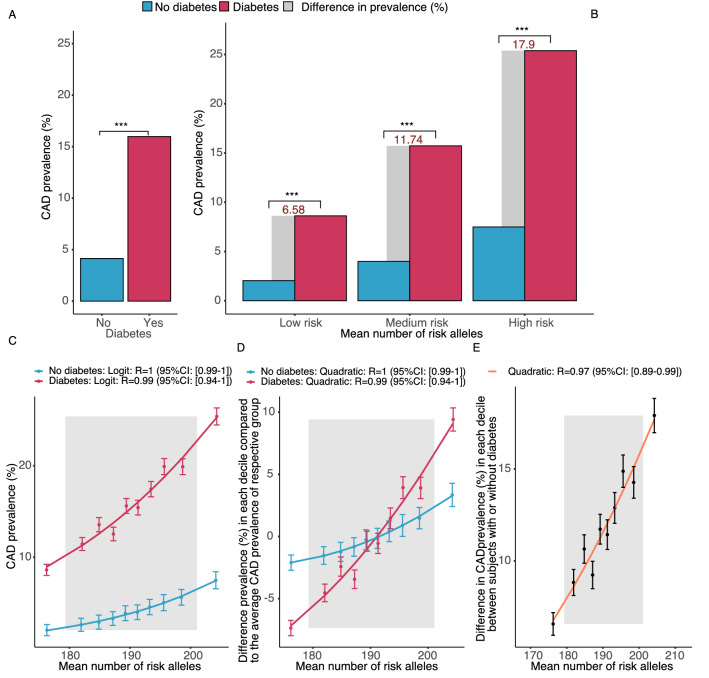
Results: In UK Biobank, risk alleles counts in the lowest (175.4) and highest decile (205.7) of the distribution differed by only 16.9%, which nevertheless increased CAD prevalence 3.4-fold (p < 0.01). Irrespective of the affected gene, a single risk allele multiplied the effects of all others carried by a person, resulting in a 2.9-fold stronger effect size in the top versus the bottom decile (p < 0.01) and an exponential increase in risk (R > 0.94). Classical risk factors shifted effect sizes to the steep upslope of the logarithmic function linking risk allele numbers with CAD prevalence. Similar phenomena were observed in the Estonian Biobank and for risk alleles affecting diabetes mellitus, breast and prostate cancer.
Conclusions: Alleles predisposing to common diseases can be carried safely in large numbers, but few additional ones lead to sharp risk increments. Here, we describe exponential functions by which risk alleles combine interchangeably but multiplicatively with each other and with modifiable risk factors to affect prevalence. Our data suggest that the biological systems underlying these diseases are modulated by hundreds of genes but become only fragile when a narrow window of total risk, irrespective of its genetic or environmental origins, has been passed.
Keywords: Coronary artery disease; Genome-wide association studies; Liability threshold; Risk prediction; Risk score.
© 2022. The Author(s).
Conflict of interest statement
The author(s) declare(s) that there is no conflict of interest.
Figures

References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
