

Scalable Bayesian phylogenetics
- PMID: 35989603
- PMCID: PMC9393558
- DOI: 10.1098/rstb.2021.0242
Scalable Bayesian phylogenetics
Abstract
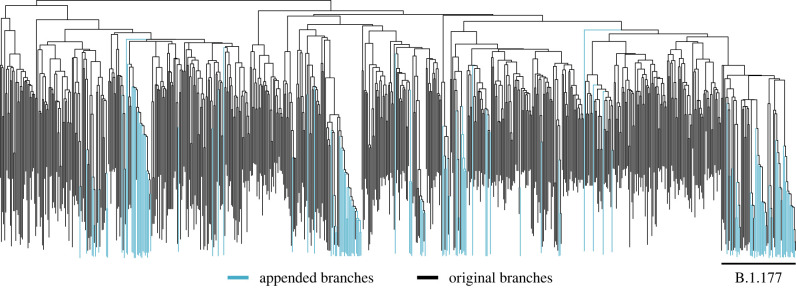
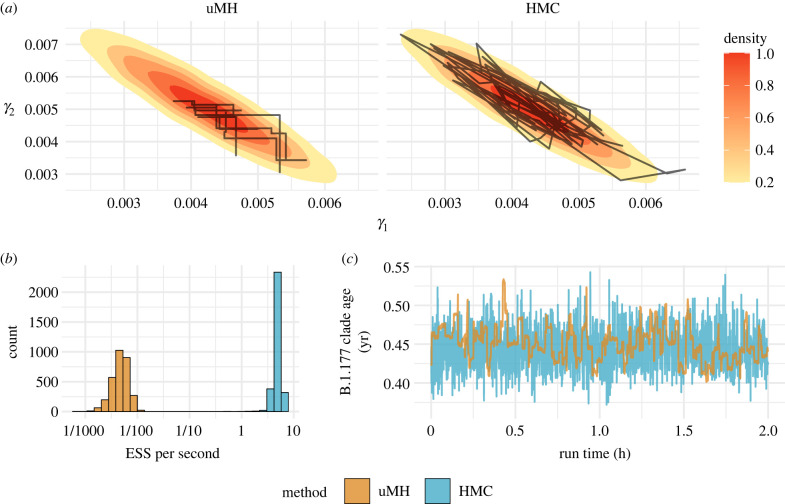
Recent advances in Bayesian phylogenetics offer substantial computational savings to accommodate increased genomic sampling that challenges traditional inference methods. In this review, we begin with a brief summary of the Bayesian phylogenetic framework, and then conceptualize a variety of methods to improve posterior approximations via Markov chain Monte Carlo (MCMC) sampling. Specifically, we discuss methods to improve the speed of likelihood calculations, reduce MCMC burn-in, and generate better MCMC proposals. We apply several of these techniques to study the evolution of HIV virulence along a 1536-tip phylogeny and estimate the internal node heights of a 1000-tip SARS-CoV-2 phylogenetic tree in order to illustrate the speed-up of such analyses using current state-of-the-art approaches. We conclude our review with a discussion of promising alternatives to MCMC that approximate the phylogenetic posterior. This article is part of a discussion meeting issue 'Genomic population structures of microbial pathogens'.
Keywords: BEAST; Bayesian phylogenetics; Hamiltonian Monte Carlo; adapative MCMC; online inference; scalable inference.
Figures
Similar articles
-
Online tree expansion could help solve the problem of scalability in Bayesian phylogenetics.Syst Biol. 2023 Nov 1;72(5):1199-1206. doi: 10.1093/sysbio/syad045. Syst Biol. 2023. PMID: 37498209 Free PMC article.
-
An Efficient Coalescent Epoch Model for Bayesian Phylogenetic Inference.Syst Biol. 2022 Oct 12;71(6):1549-1560. doi: 10.1093/sysbio/syac015. Syst Biol. 2022. PMID: 35212733 Free PMC article.
-
A comparison of computational algorithms for the Bayesian analysis of clinical trials.Clin Trials. 2024 Dec;21(6):689-700. doi: 10.1177/17407745241247334. Epub 2024 May 16. Clin Trials. 2024. PMID: 38752434 Free PMC article.
-
A biologist's guide to Bayesian phylogenetic analysis.Nat Ecol Evol. 2017 Oct;1(10):1446-1454. doi: 10.1038/s41559-017-0280-x. Epub 2017 Sep 21. Nat Ecol Evol. 2017. PMID: 28983516 Free PMC article. Review.
-
A review of the Bayesian approach with the MCMC and the HMC as a competitor of classical likelihood statistics for pharmacometricians.Transl Clin Pharmacol. 2023 Jun;31(2):69-84. doi: 10.12793/tcp.2023.31.e9. Epub 2023 Jun 26. Transl Clin Pharmacol. 2023. PMID: 37440780 Free PMC article. Review.
Cited by
-
Model design for nonparametric phylodynamic inference and applications to pathogen surveillance.Virus Evol. 2023 May 5;9(1):vead028. doi: 10.1093/ve/vead028. eCollection 2023. Virus Evol. 2023. PMID: 37229349 Free PMC article.
-
Genomic population structures of microbial pathogens.Philos Trans R Soc Lond B Biol Sci. 2022 Oct 10;377(1861):20210230. doi: 10.1098/rstb.2021.0230. Epub 2022 Aug 22. Philos Trans R Soc Lond B Biol Sci. 2022. PMID: 35989608 Free PMC article. No abstract available.
-
SARS-CoV-2 in low-income countries: the need for sustained genomic surveillance.Lancet Glob Health. 2023 Jun;11(6):e815-e816. doi: 10.1016/S2214-109X(23)00197-3. Lancet Glob Health. 2023. PMID: 37202013 Free PMC article. No abstract available.
-
Online tree expansion could help solve the problem of scalability in Bayesian phylogenetics.Syst Biol. 2023 Nov 1;72(5):1199-1206. doi: 10.1093/sysbio/syad045. Syst Biol. 2023. PMID: 37498209 Free PMC article.
-
HantaNet: A New MicrobeTrace Application for Hantavirus Classification, Genomic Surveillance, Epidemiology and Outbreak Investigations.Viruses. 2023 Nov 2;15(11):2208. doi: 10.3390/v15112208. Viruses. 2023. PMID: 38005885 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
