

Dissection of the macrophage response towards infection by the Leishmania-viral endosymbiont duo and dynamics of the type I interferon response
- PMID: 35992159
- PMCID: PMC9386148
- DOI: 10.3389/fcimb.2022.941888
Dissection of the macrophage response towards infection by the Leishmania-viral endosymbiont duo and dynamics of the type I interferon response
Abstract
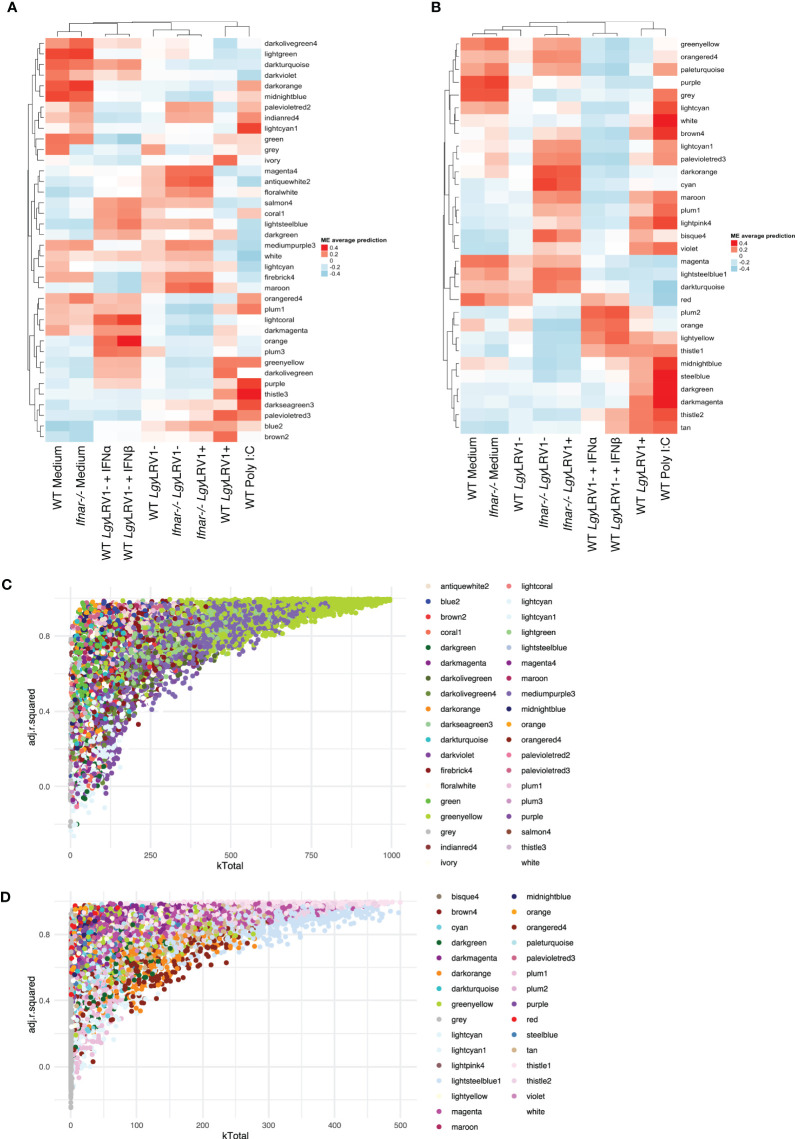
Leishmania RNA virus 1 (LRV1) is a double-stranded RNA virus found in some strains of the human protozoan parasite Leishmania, the causative agent of leishmaniasis, a neglected tropical disease. Interestingly, the presence of LRV1 inside Leishmania constitutes an important virulence factor that worsens the leishmaniasis outcome in a type I interferon (IFN)-dependent manner and contributes to treatment failure. Understanding how macrophages respond toward Leishmania alone or in combination with LRV1 as well as the role that type I IFNs may play during infection is fundamental to oversee new therapeutic strategies. To dissect the macrophage response toward infection, RNA sequencing was performed on murine wild-type and Ifnar-deficient bone marrow-derived macrophages infected with Leishmania guyanensis (Lgy) devoid or not of LRV1. Additionally, macrophages were treated with poly I:C (mimetic virus) or with type I IFNs. By implementing a weighted gene correlation network analysis, the groups of genes (modules) with similar expression patterns, for example, functionally related, coregulated, or the members of the same functional pathway, were identified. These modules followed patterns dependent on Leishmania, LRV1, or Leishmania exacerbated by the presence of LRV1. Not only the visualization of how individual genes were embedded to form modules but also how different modules were related to each other were observed. Thus, in the context of the observed hyperinflammatory phenotype associated to the presence of LRV1, it was noted that the biomarkers tumor-necrosis factor α (TNF-α) and the interleukin 6 (IL-6) belonged to different modules and that their regulating specific Src-family kinases were segregated oppositely. In addition, this network approach revealed the strong and sustained effect of LRV1 on the macrophage response and genes that had an early, late, or sustained impact during infection, uncovering the dynamics of the IFN response. Overall, this study contributed to shed light and dissect the intricate macrophage response toward infection by the Leishmania-LRV1 duo and revealed the crosstalk between modules made of coregulated genes and provided a new resource that can be further explored to study the impact of Leishmania on the macrophage response.
Keywords: Leishmania RNA virus 1 (LRV1); RNA sequencing (RNA-Seq); interleukin 6 (IL-6); macrophage; tumor-necrosis factor alpha (TNF-α); type I interferon (IFN); weighted gene coexpression network analysis (WGCNA).
Copyright © 2022 Bekkar, Isorce, Snäkä, Claudinot, Desponds, Kopelyanskiy, Prével, Reverte, Xenarios, Fasel and Teixeira.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures





References
-
- Adaui V., Lye L. F., Akopyants N. S., Zimic M., Llanos-Cuentas A., Garcia L., et al. (2016). Association of the endobiont double-stranded RNA virus LRV1 with treatment failure for human leishmaniasis caused by leishmania braziliensis in Peru and Bolivia. J. Infect. Dis. 213 (1), 112–121. doi: 10.1093/infdis/jiv354 - DOI - PMC - PubMed
-
- Alexa A., Rahnenfuhrer J. (2016). topGO: Enrichment analysis for gene ontology. r package version. Bioconductor 2(0). doi: 10.18129/B9.bioc.topGO - DOI
-
- Aoki J. I., Muxel S. M., Zampieri R.A ., Laranjeira-Silva M. F., Muller K. E., Nerland A. H, et al. (2017). RNA-Seq transcriptional profiling of leishmania amazonensis reveals an arginase-dependent gene expression regulation. PloS Negl. Trop. Dis. 11 (10), e0006026. doi: 10.1371/journal.pntd.0006026 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous