

SPEACH_AF: Sampling protein ensembles and conformational heterogeneity with Alphafold2
- PMID: 35994486
- PMCID: PMC9436118
- DOI: 10.1371/journal.pcbi.1010483
SPEACH_AF: Sampling protein ensembles and conformational heterogeneity with Alphafold2
Abstract
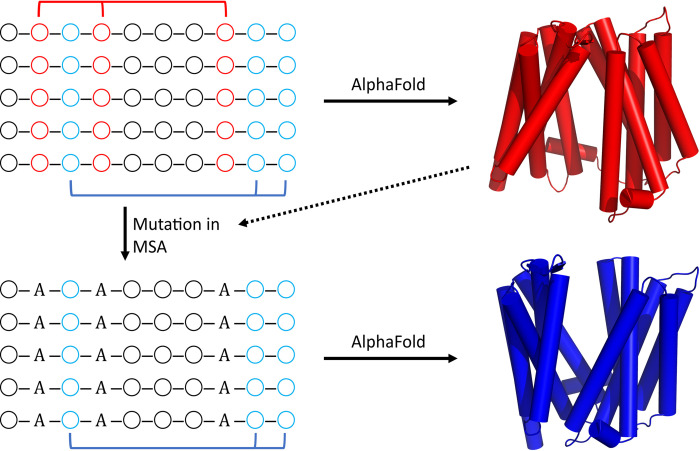
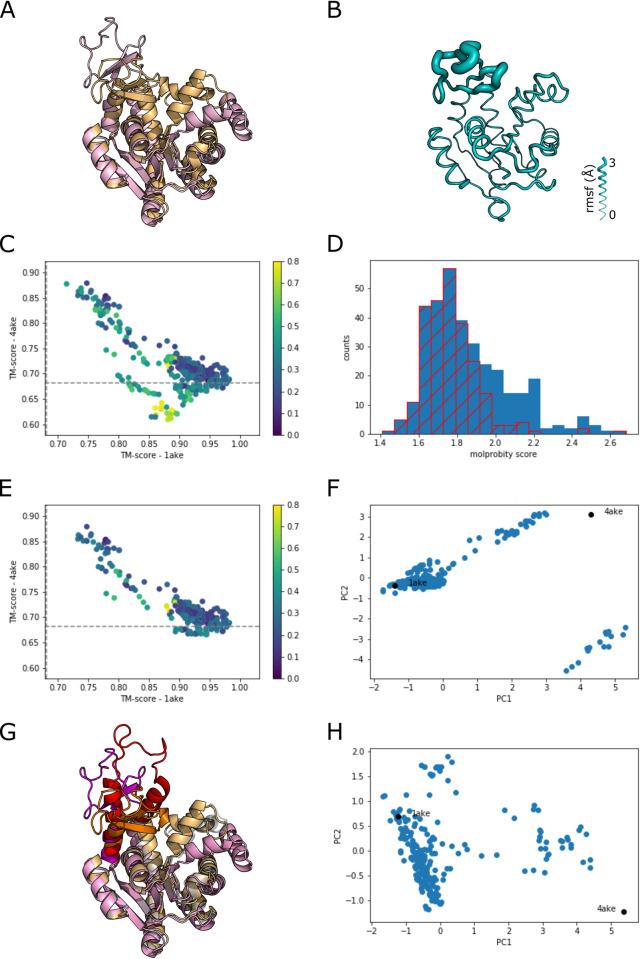
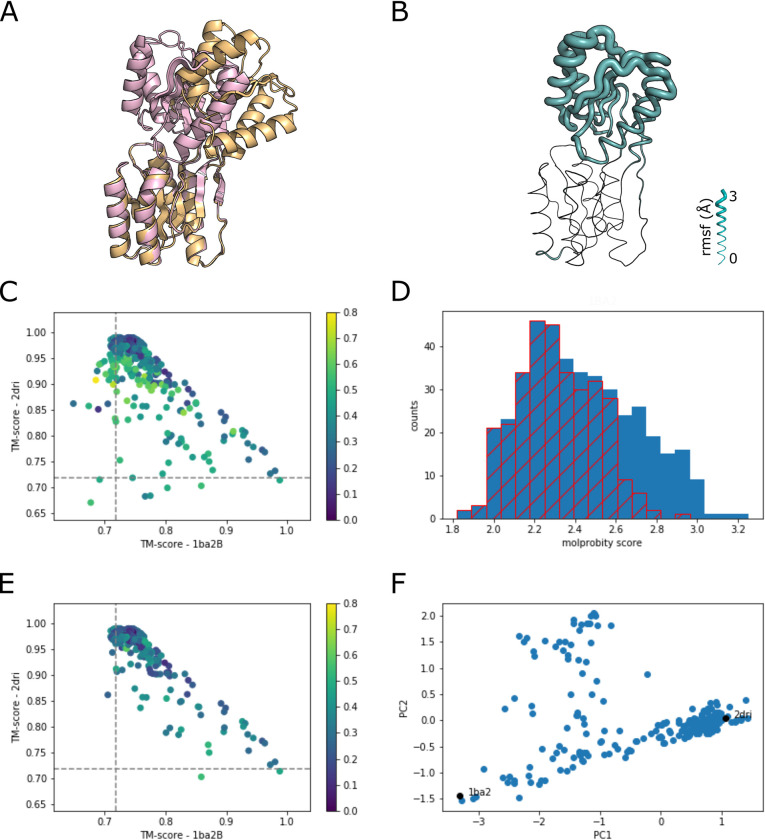
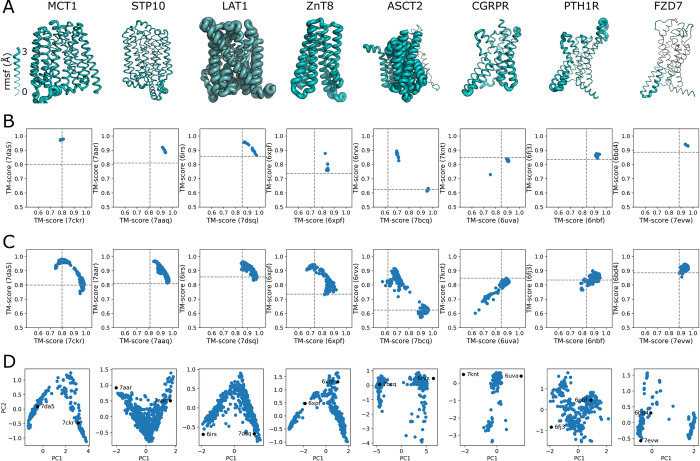
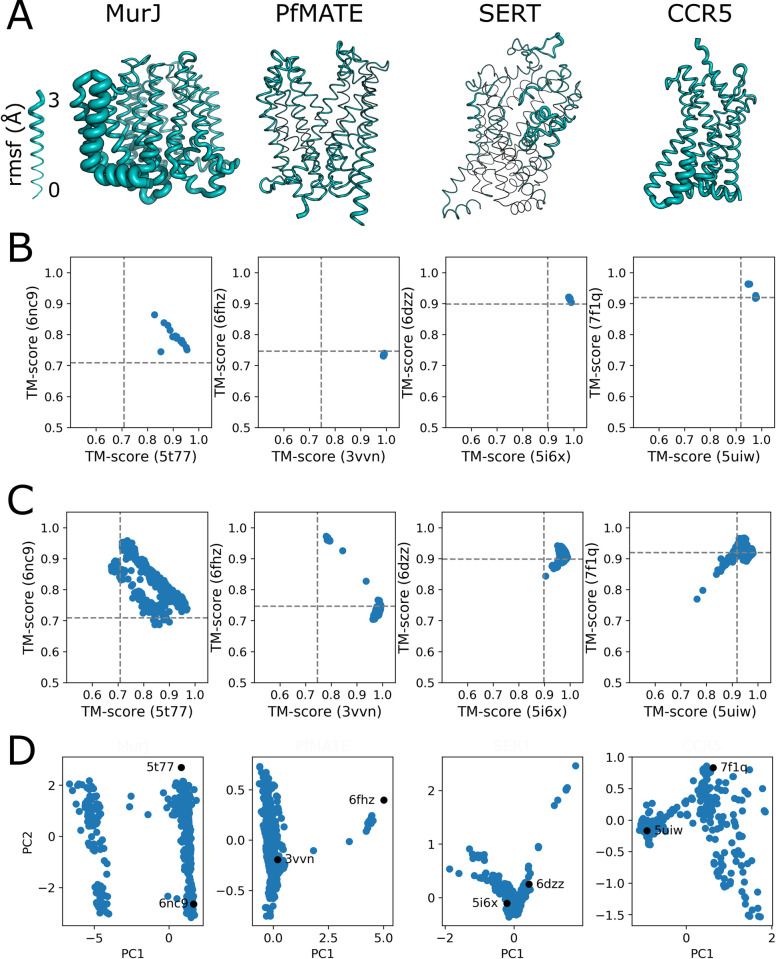
The unprecedented performance of Deepmind's Alphafold2 in predicting protein structure in CASP XIV and the creation of a database of structures for multiple proteomes and protein sequence repositories is reshaping structural biology. However, because this database returns a single structure, it brought into question Alphafold's ability to capture the intrinsic conformational flexibility of proteins. Here we present a general approach to drive Alphafold2 to model alternate protein conformations through simple manipulation of the multiple sequence alignment via in silico mutagenesis. The approach is grounded in the hypothesis that the multiple sequence alignment must also encode for protein structural heterogeneity, thus its rational manipulation will enable Alphafold2 to sample alternate conformations. A systematic modeling pipeline is benchmarked against canonical examples of protein conformational flexibility and applied to interrogate the conformational landscape of membrane proteins. This work broadens the applicability of Alphafold2 by generating multiple protein conformations to be tested biologically, biochemically, biophysically, and for use in structure-based drug design.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
