

EGR1 Haploinsufficiency Confers a Fitness Advantage to Hematopoietic Stem Cells Following Chemotherapy
- PMID: 35995095
- PMCID: PMC10617250
- DOI: 10.1016/j.exphem.2022.08.003
EGR1 Haploinsufficiency Confers a Fitness Advantage to Hematopoietic Stem Cells Following Chemotherapy
Abstract
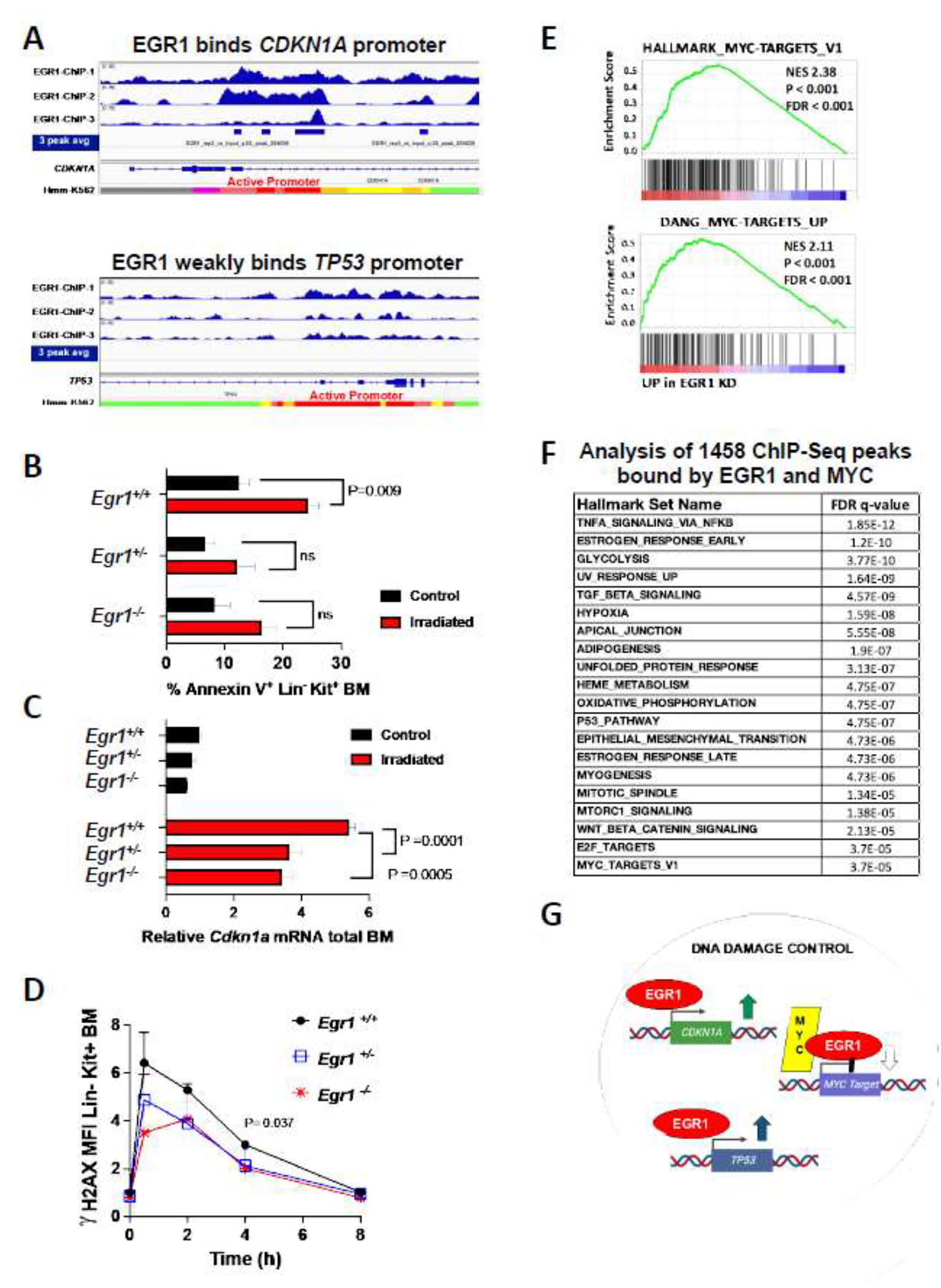
Therapy-related myeloid neoplasms (t-MNs) share many clinical and molecular characteristics with AML de novo in the elderly. One common factor is that they arise in the setting of chronic inflammation, likely because of advanced age or chemotherapy-induced senescence. Here, we examined the effect of haploinsufficient loss of the del(5q) tumor suppressor gene, EGR1, commonly deleted in high-risk MNs. In mice, under the exogenous stress of either serial transplant or successive doses of the alkylating agent N-ethyl-nitrosourea (ENU), Egr1-haploinsufficient hematopoietic stem cells (HSCs) exhibit a clonal advantage. Complete loss of EGR1 function is incompatible with transformation; mutations of EGR1 are rare and are not observed in the remaining allele in del(5q) patients, and complete knockout of Egr1 in mice leads to HSC exhaustion. Using chromatin immunoprecipitation sequencing (ChIP-seq), we identified EGR1 binding sites in human CD34+ cord blood-derived stem and progenitor cells (HSPCs) and found that EGR1 binds genes critical for stem cell differentiation, inflammatory signaling, and the DNA damage response. Notably, in the chromosome 5 sequences frequently deleted in patients, there is a significant enrichment of innate and inflammatory genes, which may confer a fitness advantage in an inflammatory environment. Short hairpin RNA (shRNA)-mediated silencing of EGR1 biases HSPCs toward a self-renewal transcriptional signature. In the absence of EGR1, HSPCs are characterized by upregulated MYC-driven proliferative signals, downregulated CDKN1A (p21), disrupted DNA damage response, and downregulated inflammation-adaptations anticipated to confer a relative fitness advantage for stem cells especially in an environment of chronic inflammation.
Copyright © 2022 ISEH -- Society for Hematology and Stem Cells. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest disclosure The authors declare no potential conflicts of interest.
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
