

Association mapping across a multitude of traits collected in diverse environments in maize
- PMID: 35997208
- PMCID: PMC9396454
- DOI: 10.1093/gigascience/giac080
Association mapping across a multitude of traits collected in diverse environments in maize
Abstract
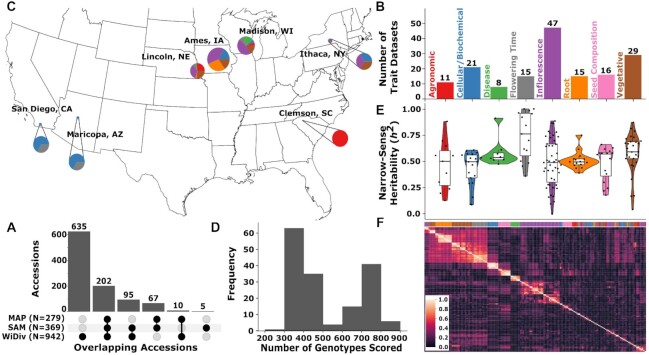
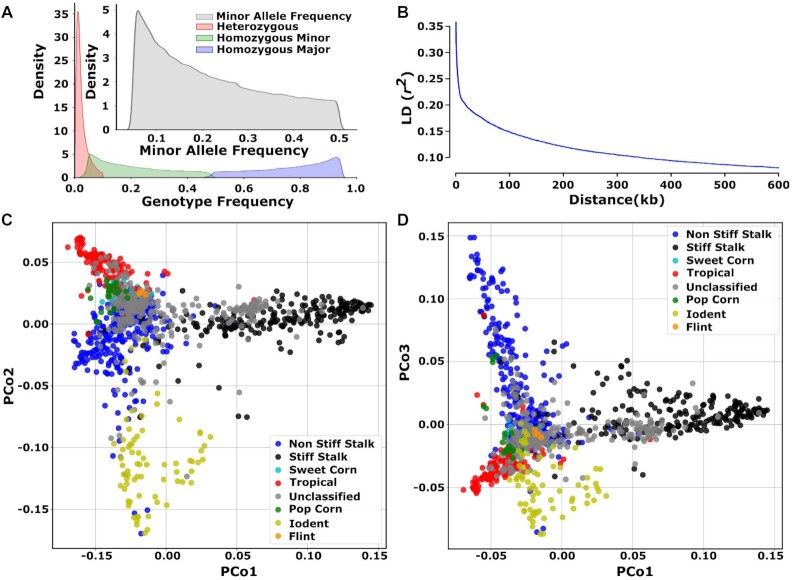
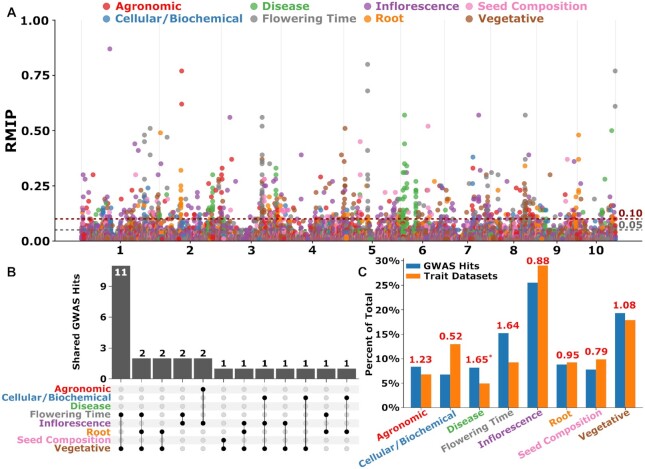
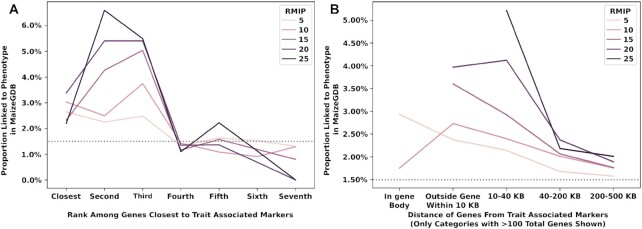
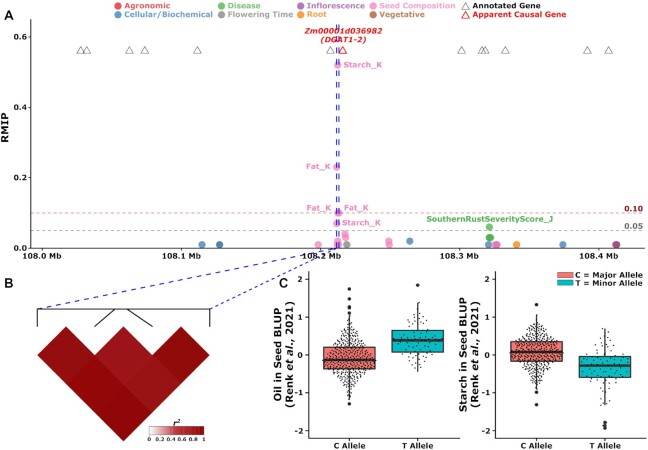
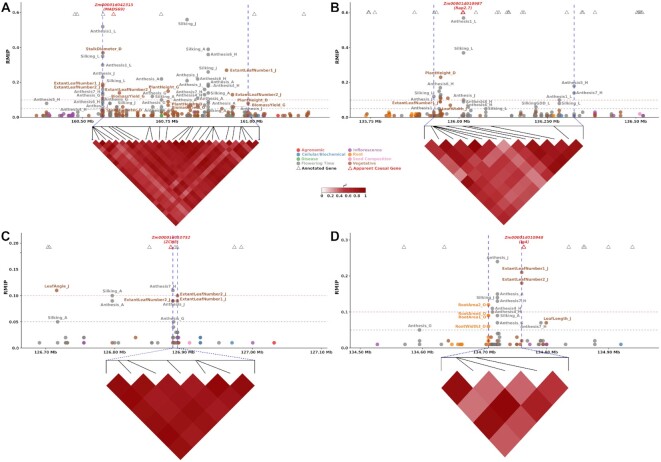
Classical genetic studies have identified many cases of pleiotropy where mutations in individual genes alter many different phenotypes. Quantitative genetic studies of natural genetic variants frequently examine one or a few traits, limiting their potential to identify pleiotropic effects of natural genetic variants. Widely adopted community association panels have been employed by plant genetics communities to study the genetic basis of naturally occurring phenotypic variation in a wide range of traits. High-density genetic marker data-18M markers-from 2 partially overlapping maize association panels comprising 1,014 unique genotypes grown in field trials across at least 7 US states and scored for 162 distinct trait data sets enabled the identification of of 2,154 suggestive marker-trait associations and 697 confident associations in the maize genome using a resampling-based genome-wide association strategy. The precision of individual marker-trait associations was estimated to be 3 genes based on a reference set of genes with known phenotypes. Examples were observed of both genetic loci associated with variation in diverse traits (e.g., above-ground and below-ground traits), as well as individual loci associated with the same or similar traits across diverse environments. Many significant signals are located near genes whose functions were previously entirely unknown or estimated purely via functional data on homologs. This study demonstrates the potential of mining community association panel data using new higher-density genetic marker sets combined with resampling-based genome-wide association tests to develop testable hypotheses about gene functions, identify potential pleiotropic effects of natural genetic variants, and study genotype-by-environment interaction.
Keywords: community association populations; maize; pleiotropy; quantitative genetics.
© The Author(s) 2022. Published by Oxford University Press GigaScience.
Conflict of interest statement
J.C.S. has equity interests in Data2Bio, LLC; Dryland Genetics LLC; and EnGeniousAg LLC. He is a member of the scientific advisory board of GeneSeek and currently serves as a guest editor for
Figures
References
-
- Flint-Garcia SA, Thuillet AC, Yu J, et al. Maize association population: a high-resolution platform for quantitative trait locus dissection. Plant J. 2005;44(6):1054–64. - PubMed
-
- Hansey CN, Johnson JM, Sekhon RS, et al. Genetic diversity of a maize association population with restricted phenology. Crop Sci. 2011;51(2):704–15.
-
- Yang X, Gao S, Xu S, et al. Characterization of a global germplasm collection and its potential utilization for analysis of complex quantitative traits in maize. Mol Breeding. 2011;28(4):511–26.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
