

Cleft Palate in Apert Syndrome
- PMID: 35997397
- PMCID: PMC9397066
- DOI: 10.3390/jdb10030033
Cleft Palate in Apert Syndrome
Abstract
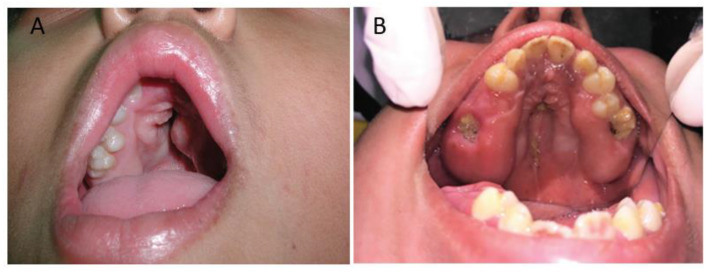
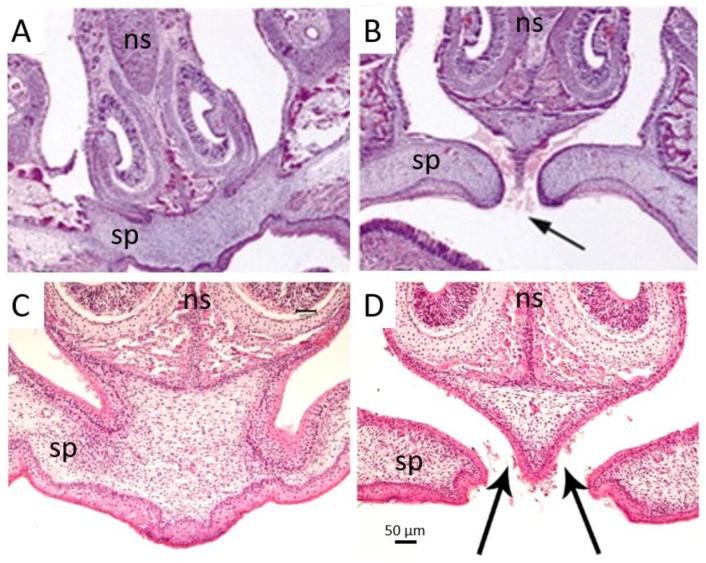
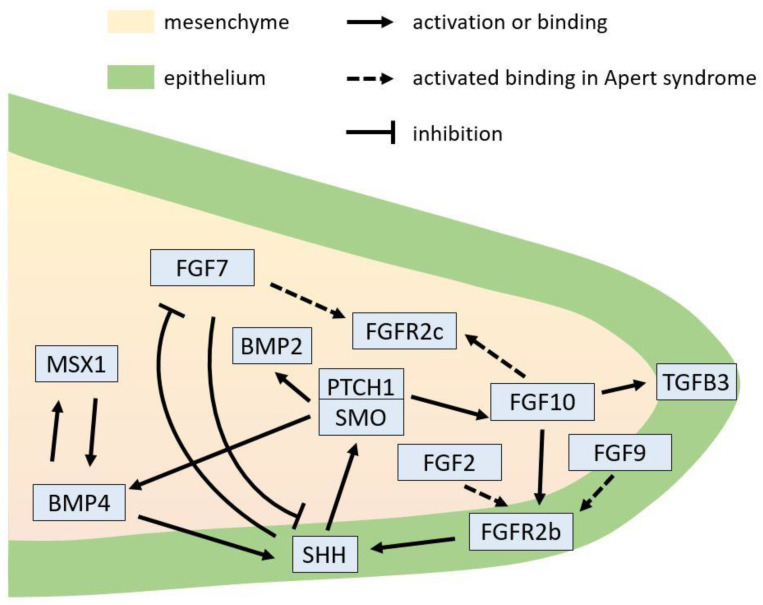
Apert syndrome is a rare genetic disorder characterized by craniosynostosis, midface retrusion, and limb anomalies. Cleft palate occurs in a subset of Apert syndrome patients. Although the genetic causes underlying Apert syndrome have been identified, the downstream signaling pathways and cellular mechanisms responsible for cleft palate are still elusive. To find clues for the pathogenic mechanisms of palatal defects in Apert syndrome, we review the clinical characteristics of the palate in cases of Apert syndrome, the palatal phenotypes in mouse models, and the potential signaling mechanisms involved in palatal defects. In Apert syndrome patients, cleft of the soft palate is more frequent than of the hard palate. The length of the hard palate is decreased. Cleft palate is associated most commonly with the S252W variant of FGFR2. In addition to cleft palate, high-arched palate, lateral palatal swelling, or bifid uvula are common in Apert syndrome patients. Mouse models of Apert syndrome display palatal defects, providing valuable tools to understand the underlying mechanisms. The mutations in FGFR2 causing Apert syndrome may change a signaling network in epithelial-mesenchymal interactions during palatogenesis. Understanding the pathogenic mechanisms of palatal defects in Apert syndrome may shed light on potential novel therapeutic solutions.
Keywords: Apert syndrome; FGF; FGFR2; cleft palate; high-arched palate; palatogenesis; pseudo-cleft palate; uvula.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Mai C.T., Cassell C.H., Meyer R.E., Isenburg J., Canfield M.A., Rickard R., Olney R.S., Stallings E.B., Beck M., Hashmi S.S., et al. Birth defects data from population-based birth defects surveillance programs in the United States, 2007 to 2011: Highlighting orofacial clefts. Birth Defects Res. Part A Clin. Mol. Teratol. 2014;100:895–904. doi: 10.1002/bdra.23329. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
