

Mycobacterium tuberculosis Utilizes Host Histamine Receptor H1 to Modulate Reactive Oxygen Species Production and Phagosome Maturation via the p38MAPK-NOX2 Axis
- PMID: 36000734
- PMCID: PMC9600773
- DOI: 10.1128/mbio.02004-22
Mycobacterium tuberculosis Utilizes Host Histamine Receptor H1 to Modulate Reactive Oxygen Species Production and Phagosome Maturation via the p38MAPK-NOX2 Axis
Abstract
Tuberculosis (TB), which is caused by the single pathogenic bacterium, Mycobacterium tuberculosis, is among the top 10 lethal diseases worldwide. This situation has been exacerbated by the increasing number of cases of multidrug-resistant TB (MDR-TB) and extensively drug-resistant TB (XDR-TB). Histamine is an organic nitrogenous compound that mediates a plethora of cell processes via different receptors. The expression of histamine receptor H1 (HRH1), one of the four histamine receptors identified to date was previously reported to be augmented by M. tuberculosis infection, although the underlying mechanism is unclear. In the present study, we applied confocal microscopy, flow cytometry, and Western blotting to show that HRH1 expression was enhanced in macrophages following mycobacterial infection. Furthermore, by combining techniques of gene knockdown, immunoprecipitation, intracellular bacterial burden analysis, fluorescence labeling, and imaging, we found that M. tuberculosis targeted the host HRH1 to suppress NOX2-mediated cROS production and inhibit phagosome maturation and acidification via the GRK2-p38MAPK signaling pathway. Our findings clarified the underlying mechanism of the M. tuberculosis and host HRH1 interaction and may provide useful information for the development of novel antituberculosis treatments. IMPORTANCE Once engulfed in macrophage phagosomes, M. tuberculosis adopts various strategies to take advantage of the host environment for its intracellular survival. Histamine is an organic nitrogen-containing compound that mediates a plethora of cellular processes via different receptors, but the crosstalk mechanism between M. tuberculosis and HRH1 in macrophages is not clear. Our results revealed that M. tuberculosis infection enhanced HRH1 expression, which in turn restrained macrophage bactericidal activity by modulating the GRK2-p38MAPK signaling pathway, inhibiting NOX2-mediated cROS production and phagosome maturation. Clarification of the underlying mechanism by which M. tuberculosis utilizes host HRH1 to favor its intracellular survival may provide useful information for the development of novel antituberculosis treatments.
Keywords: HRH1; Mycobacterium tuberculosis; NOX2; ROS; p38MAPK.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



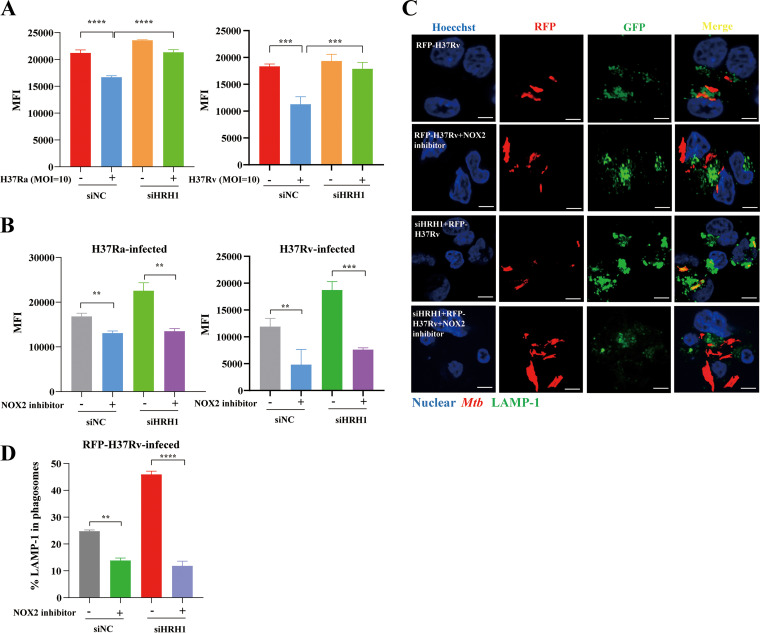
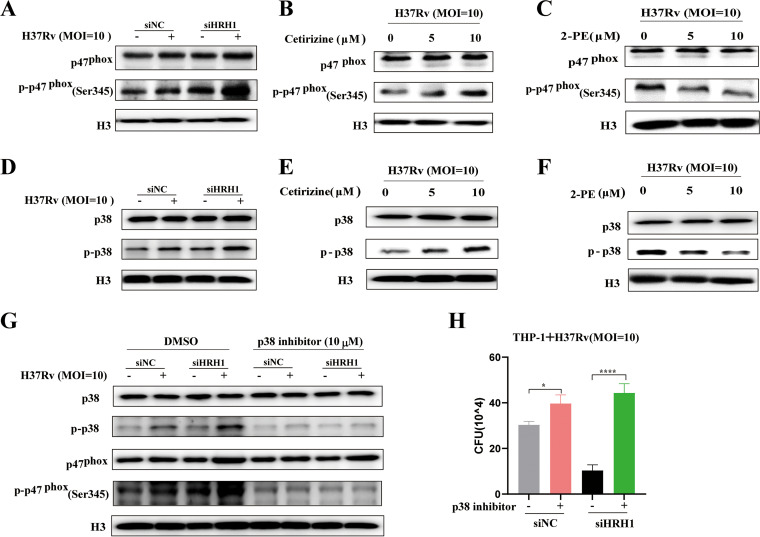
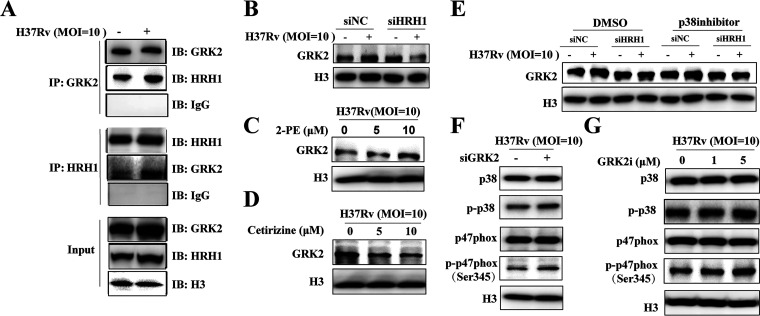

Similar articles
-
Lysophosphatidylcholine Promotes Phagosome Maturation and Regulates Inflammatory Mediator Production Through the Protein Kinase A-Phosphatidylinositol 3 Kinase-p38 Mitogen-Activated Protein Kinase Signaling Pathway During Mycobacterium tuberculosis Infection in Mouse Macrophages.Front Immunol. 2018 Apr 27;9:920. doi: 10.3389/fimmu.2018.00920. eCollection 2018. Front Immunol. 2018. PMID: 29755479 Free PMC article.
-
[Frontier of mycobacterium research--host vs. mycobacterium].Kekkaku. 2005 Sep;80(9):613-29. Kekkaku. 2005. PMID: 16245793 Japanese.
-
CD157 Confers Host Resistance to Mycobacterium tuberculosis via TLR2-CD157-PKCzeta-Induced Reactive Oxygen Species Production.mBio. 2019 Aug 27;10(4):e01949-19. doi: 10.1128/mBio.01949-19. mBio. 2019. PMID: 31455656 Free PMC article.
-
[Development of antituberculous drugs: current status and future prospects].Kekkaku. 2006 Dec;81(12):753-74. Kekkaku. 2006. PMID: 17240921 Review. Japanese.
-
Cell biology of mycobacterium tuberculosis phagosome.Annu Rev Cell Dev Biol. 2004;20:367-94. doi: 10.1146/annurev.cellbio.20.010403.114015. Annu Rev Cell Dev Biol. 2004. PMID: 15473845 Review.
Cited by
-
Mycobacterium tuberculosis-macrophage interaction: Molecular updates.Front Cell Infect Microbiol. 2023 Mar 3;13:1062963. doi: 10.3389/fcimb.2023.1062963. eCollection 2023. Front Cell Infect Microbiol. 2023. PMID: 36936766 Free PMC article. Review.
-
Exploring host-pathogen interactions in the Dictyostelium discoideum-Mycobacterium marinum infection model of tuberculosis.Dis Model Mech. 2024 Jul 1;17(7):dmm050698. doi: 10.1242/dmm.050698. Epub 2024 Jul 22. Dis Model Mech. 2024. PMID: 39037280 Free PMC article. Review.
-
The Parkinson's drug benztropine possesses histamine receptor 1-dependent host-directed antimicrobial activity against Mycobacterium tuberculosis.NPJ Antimicrob Resist. 2025 Aug 4;3(1):70. doi: 10.1038/s44259-025-00143-x. NPJ Antimicrob Resist. 2025. PMID: 40760081 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
