

Divergent genomic trajectories predate the origin of animals and fungi
- PMID: 36002568
- PMCID: PMC9492541
- DOI: 10.1038/s41586-022-05110-4
Divergent genomic trajectories predate the origin of animals and fungi
Abstract
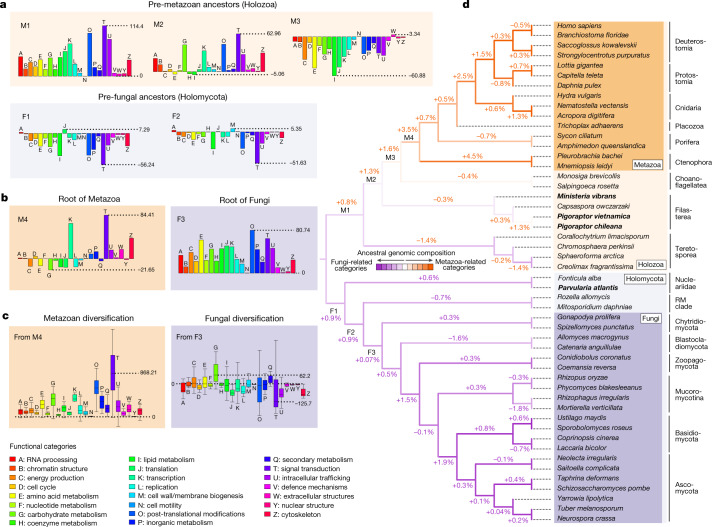
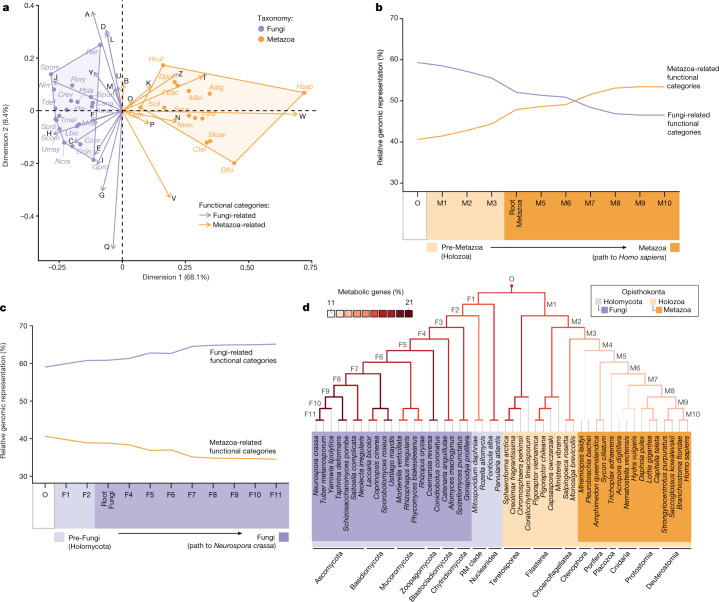
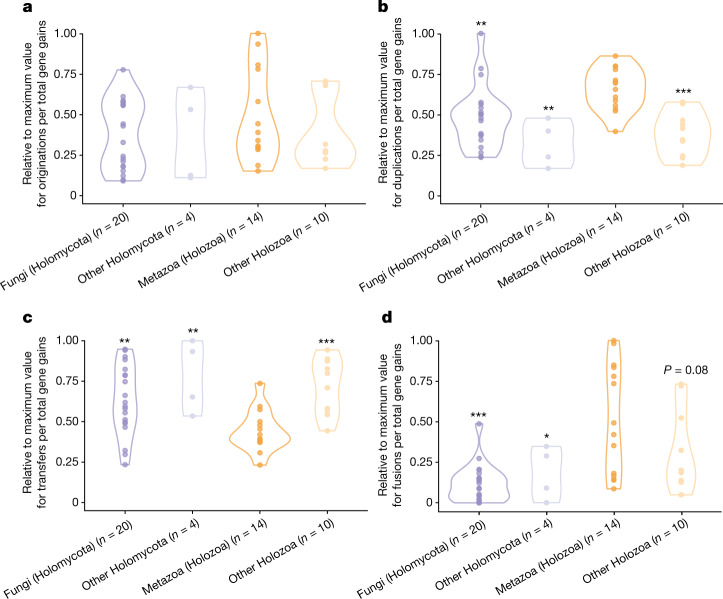
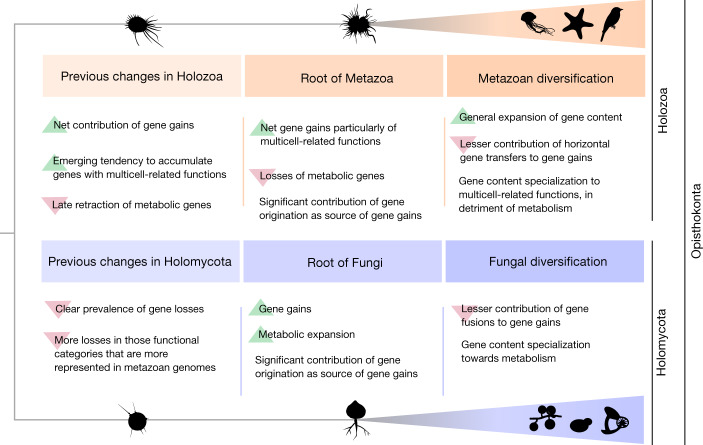
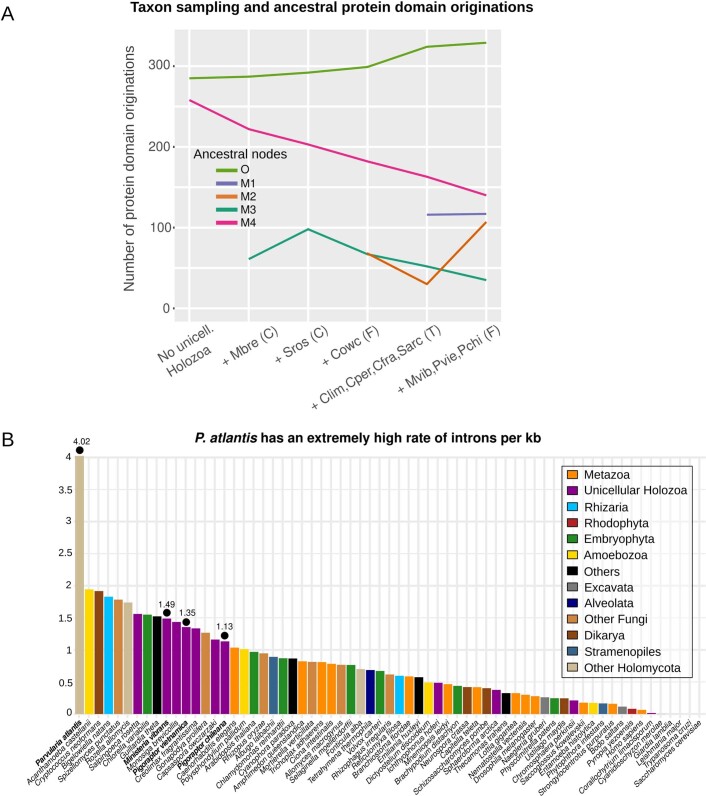
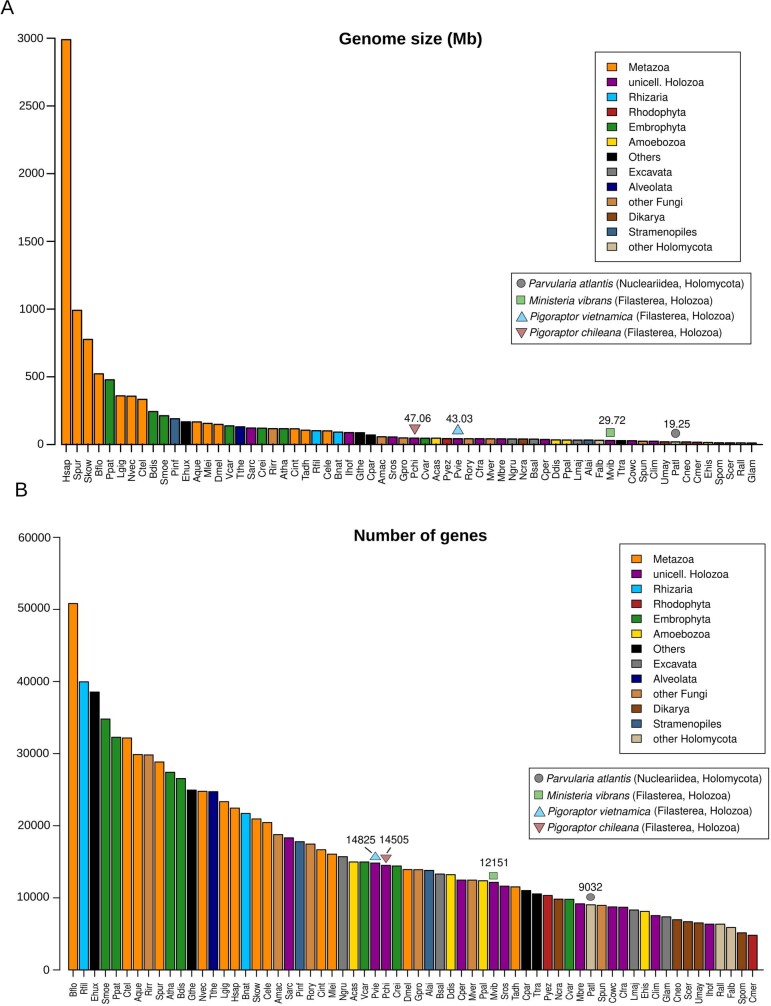
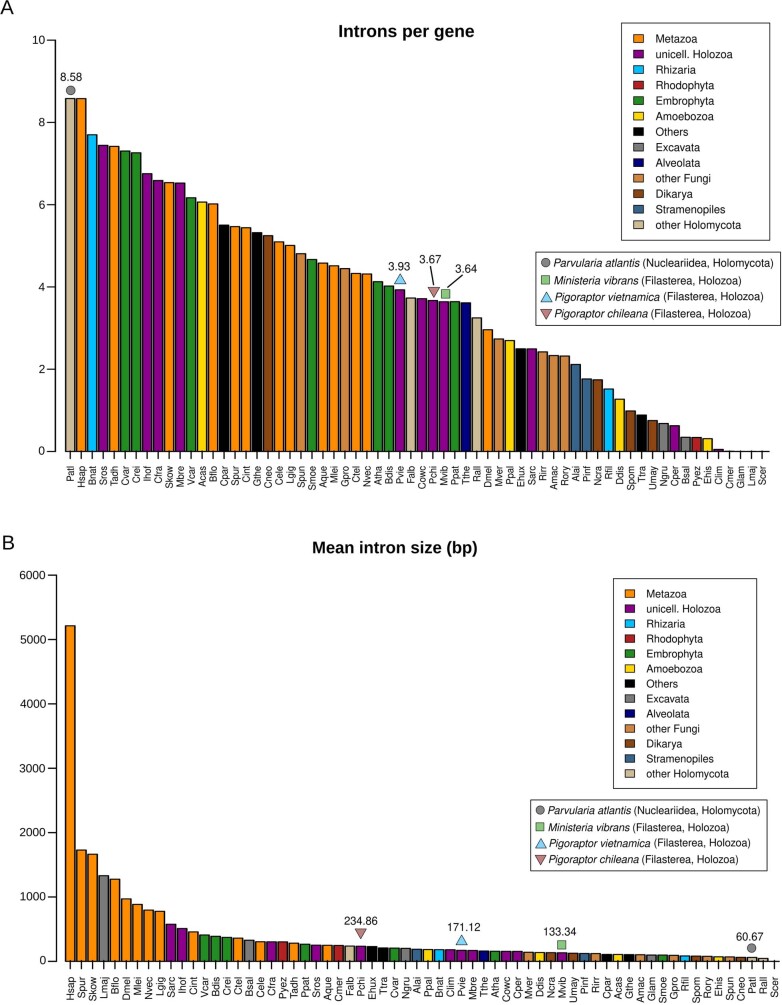
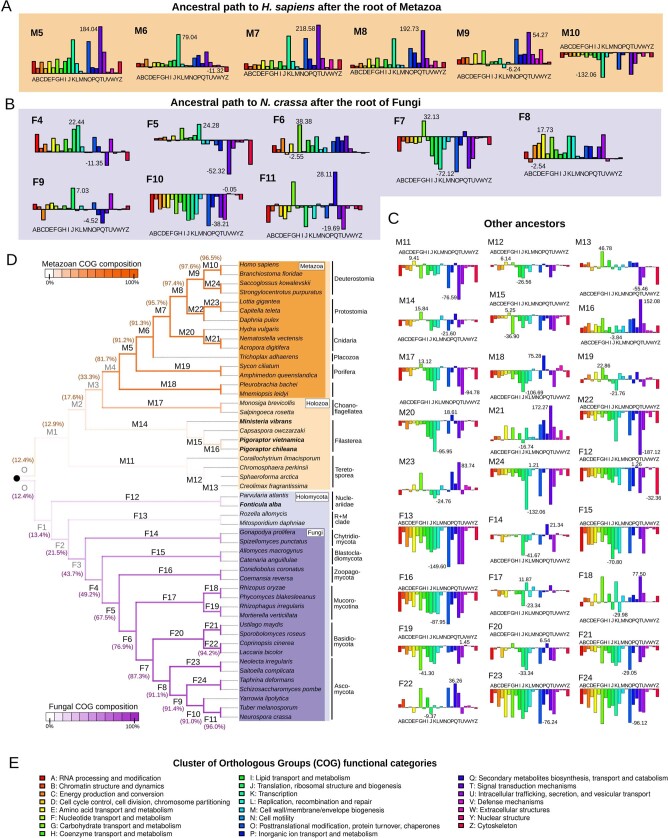
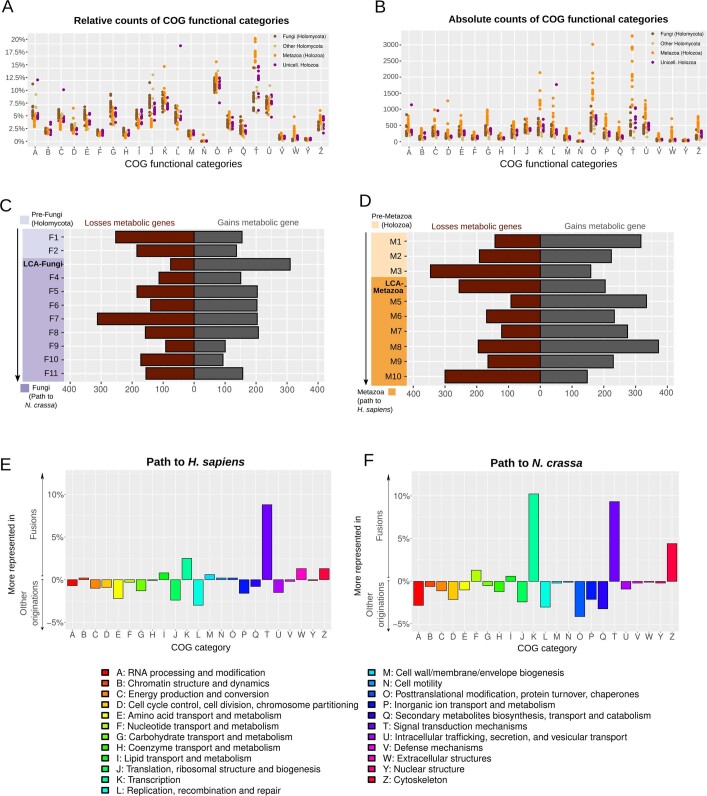
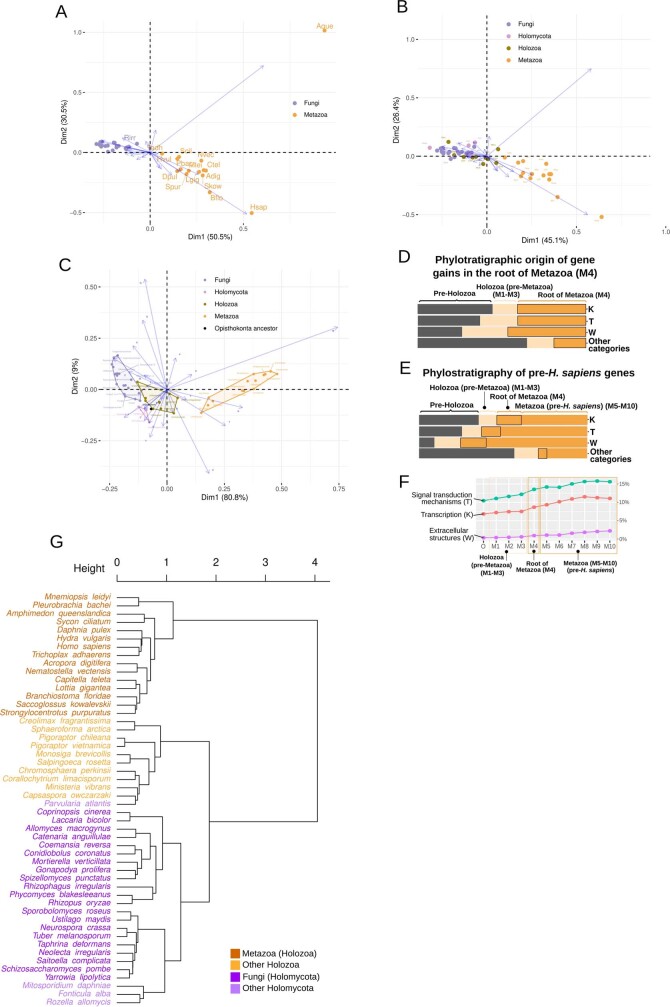
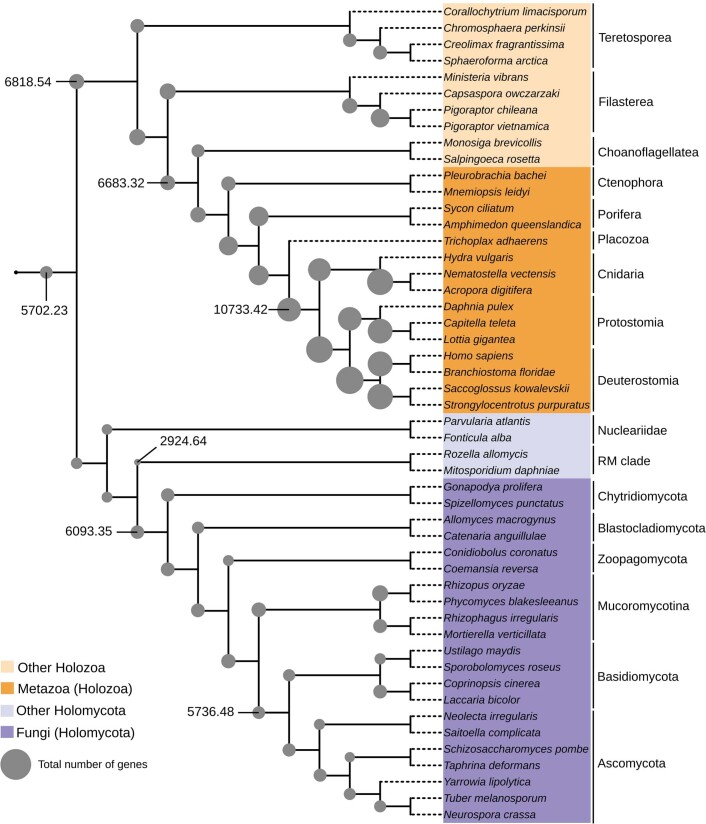
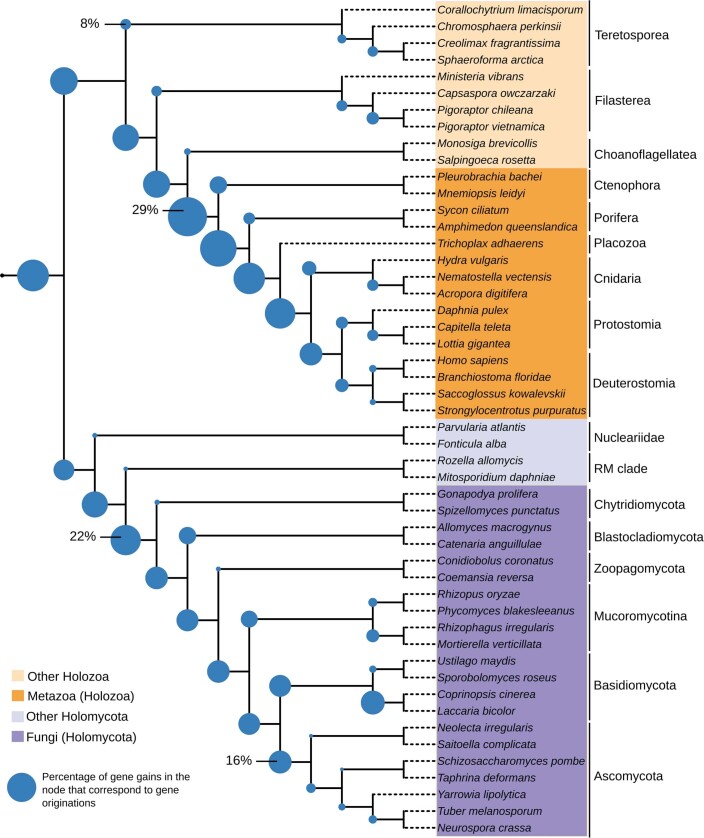
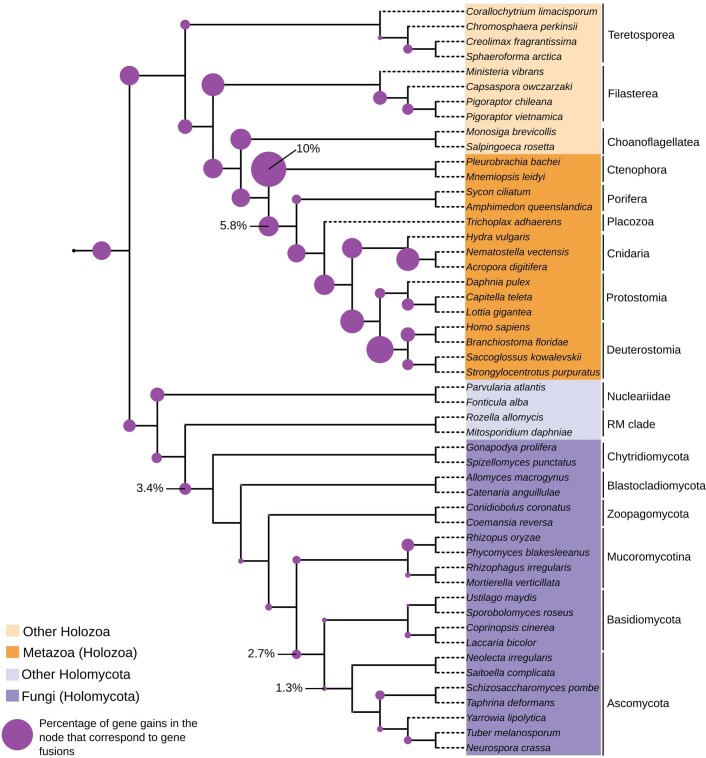
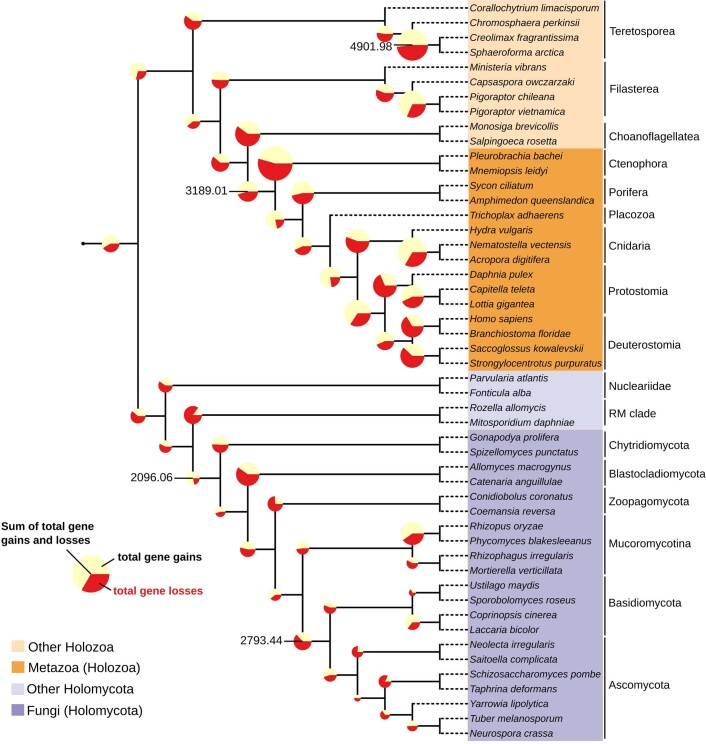
Animals and fungi have radically distinct morphologies, yet both evolved within the same eukaryotic supergroup: Opisthokonta1,2. Here we reconstructed the trajectory of genetic changes that accompanied the origin of Metazoa and Fungi since the divergence of Opisthokonta with a dataset that includes four novel genomes from crucial positions in the Opisthokonta phylogeny. We show that animals arose only after the accumulation of genes functionally important for their multicellularity, a tendency that began in the pre-metazoan ancestors and later accelerated in the metazoan root. By contrast, the pre-fungal ancestors experienced net losses of most functional categories, including those gained in the path to Metazoa. On a broad-scale functional level, fungal genomes contain a higher proportion of metabolic genes and diverged less from the last common ancestor of Opisthokonta than did the gene repertoires of Metazoa. Metazoa and Fungi also show differences regarding gene gain mechanisms. Gene fusions are more prevalent in Metazoa, whereas a larger fraction of gene gains were detected as horizontal gene transfers in Fungi and protists, in agreement with the long-standing idea that transfers would be less relevant in Metazoa due to germline isolation3-5. Together, our results indicate that animals and fungi evolved under two contrasting trajectories of genetic change that predated the origin of both groups. The gradual establishment of two clearly differentiated genomic contexts thus set the stage for the emergence of Metazoa and Fungi.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
The origin of animals and fungi.Nat Rev Genet. 2022 Nov;23(11):648-649. doi: 10.1038/s41576-022-00533-1. Nat Rev Genet. 2022. PMID: 36075981 No abstract available.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
