

In Vivo Clonal Analysis Reveals Random Monoallelic Expression in Lymphocytes That Traces Back to Hematopoietic Stem Cells
- PMID: 36003148
- PMCID: PMC9393635
- DOI: 10.3389/fcell.2022.827774
In Vivo Clonal Analysis Reveals Random Monoallelic Expression in Lymphocytes That Traces Back to Hematopoietic Stem Cells
Abstract
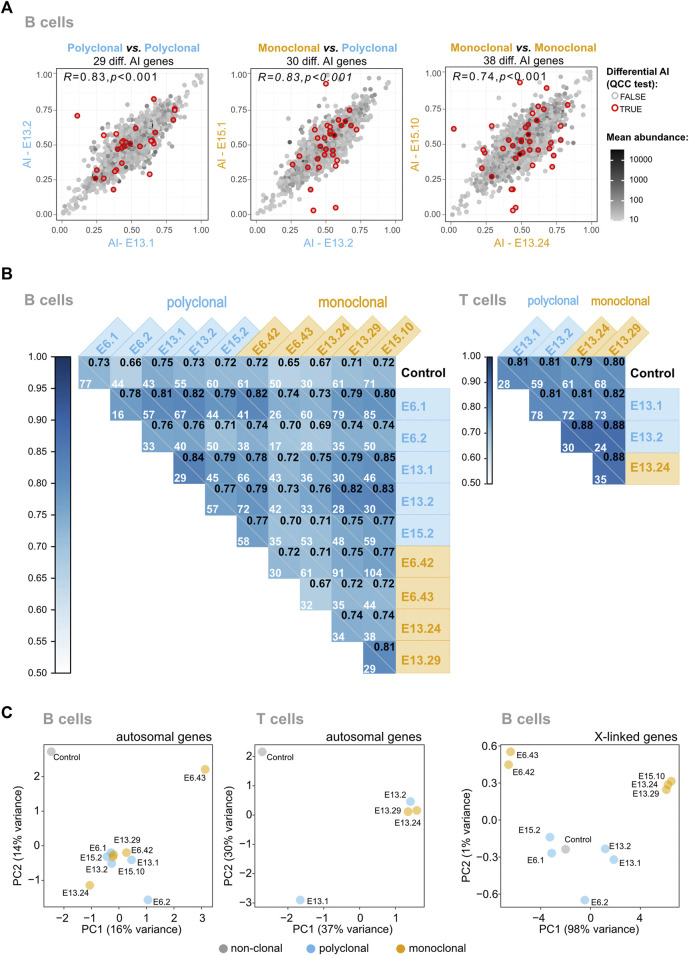
Evaluating the epigenetic landscape in the stem cell compartment at the single-cell level is essential to assess the cells' heterogeneity and predict their fate. Here, using a genome-wide transcriptomics approach in vivo, we evaluated the allelic expression imbalance in the progeny of single hematopoietic cells (HSCs) as a read-out of epigenetic marking. After 4 months of extensive proliferation and differentiation, we found that X-chromosome inactivation (XCI) is tightly maintained in all single-HSC derived hematopoietic cells. In contrast, the vast majority of the autosomal genes did not show clonal patterns of random monoallelic expression (RME). However, a persistent allele-specific autosomal transcription in HSCs and their progeny was found in a rare number of cases, none of which has been previously reported. These data show that: 1) XCI and RME in the autosomal chromosomes are driven by different mechanisms; 2) the previously reported high frequency of genes under RME in clones expanded in vitro (up to 15%) is not found in clones undergoing multiple differentiation steps in vivo; 3) prior to differentiation, HSCs have stable patterns of autosomal RME. We propose that most RME patterns in autosomal chromosomes are erased and established de novo during cell lineage differentiation.
Keywords: RNA-seq; X-chromosome inactivation (XCI); allele-specific expression; allelic imbalance (AI); clonal analysis; epigenetics; hematopoietic stem cell (HSC); random monoallelic expression (RME).
Copyright © 2022 Kubasova, Alves-Pereira, Gupta, Vinogradova, Gimelbrant and Barreto.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures





Similar articles
-
Random monoallelic expression of genes on autosomes: Parallels with X-chromosome inactivation.Semin Cell Dev Biol. 2016 Aug;56:100-110. doi: 10.1016/j.semcdb.2016.04.007. Epub 2016 Apr 19. Semin Cell Dev Biol. 2016. PMID: 27101886 Review.
-
Developmental dynamics and disease potential of random monoallelic gene expression.Dev Cell. 2014 Feb 24;28(4):366-80. doi: 10.1016/j.devcel.2014.01.016. Dev Cell. 2014. PMID: 24576422
-
Binary outcomes of enhancer activity underlie stable random monoallelic expression.Elife. 2022 May 26;11:e74204. doi: 10.7554/eLife.74204. Elife. 2022. PMID: 35617021 Free PMC article.
-
Autosomal monoallelic expression in the mouse.Genome Biol. 2012 Feb 20;13(2):R10. doi: 10.1186/gb-2012-13-2-r10. Genome Biol. 2012. PMID: 22348269 Free PMC article.
-
X chromosome inactivation in human pluripotent stem cells as a model for human development: back to the drawing board?Hum Reprod Update. 2017 Sep 1;23(5):520-532. doi: 10.1093/humupd/dmx015. Hum Reprod Update. 2017. PMID: 28582519 Review.
References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases