

The underlying pathological mechanism of ferroptosis in the development of cardiovascular disease
- PMID: 36003910
- PMCID: PMC9393259
- DOI: 10.3389/fcvm.2022.964034
The underlying pathological mechanism of ferroptosis in the development of cardiovascular disease
Abstract
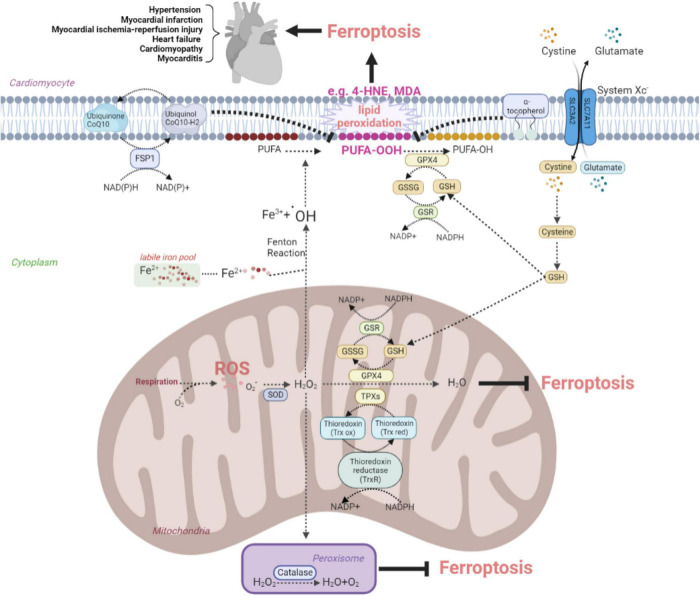
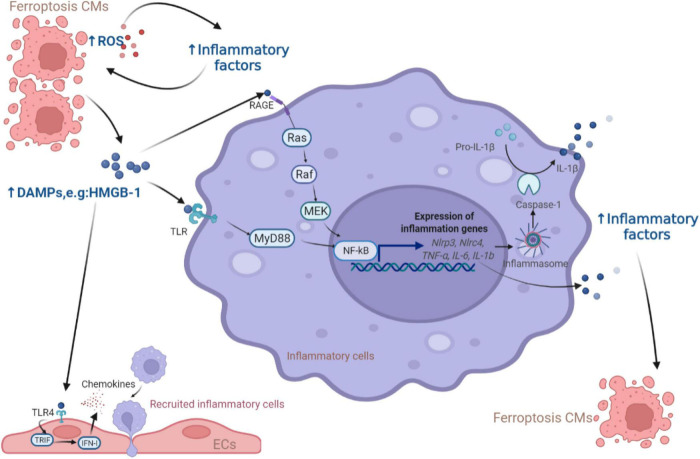
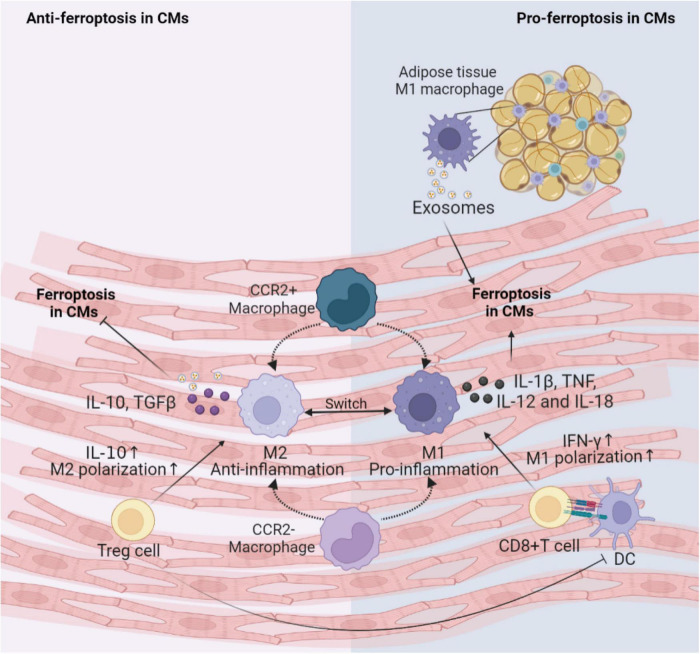
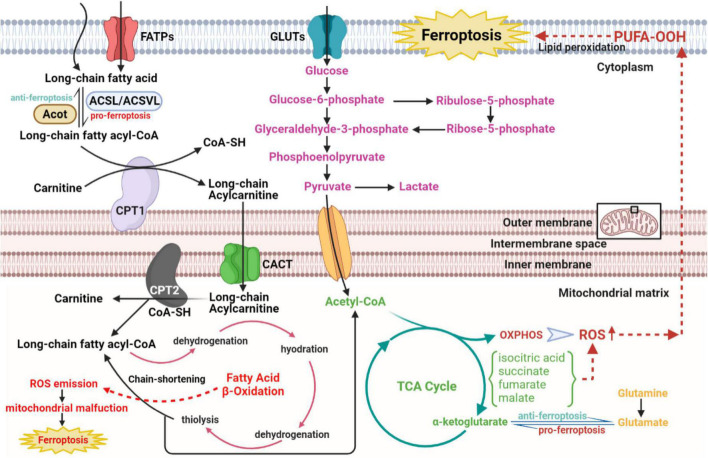
Cardiovascular diseases (CVDs) have been attracting the attention of academic society for decades. Numerous researchers contributed to figuring out the core mechanisms underlying CVDs. Among those, pathological decompensated cellular loss posed by cell death in different kinds, namely necrosis, apoptosis and necroptosis, was widely regarded to accelerate the pathological development of most heart diseases and deteriorate cardiac function. Recently, apart from programmed cell death revealed previously, ferroptosis, a brand-new cellular death identified by its ferrous-iron-dependent manner, has been demonstrated to govern the occurrence and development of different cardiovascular disorders in many types of research as well. Therefore, clarifying the regulatory function of ferroptosis is conducive to finding out strategies for cardio-protection in different conditions and improving the prognosis of CVDs. Here, molecular mechanisms concerned are summarized systematically and categorized to depict the regulatory network of ferroptosis and point out potential therapeutic targets for diverse cardiovascular disorders.
Keywords: cardiovascular disease; ferroptosis; mechanism; metabolism; pathology.
Copyright © 2022 Zhang, Tang and Yang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Targeting ferroptosis and ferritinophagy: new targets for cardiovascular diseases.J Zhejiang Univ Sci B. 2024 Jan 15;25(1):1-22. doi: 10.1631/jzus.B2300097. J Zhejiang Univ Sci B. 2024. PMID: 38163663 Free PMC article. Review.
-
Emerging roles of ferroptosis in cardiovascular diseases.Cell Death Discov. 2022 Sep 20;8(1):394. doi: 10.1038/s41420-022-01183-2. Cell Death Discov. 2022. PMID: 36127318 Free PMC article. Review.
-
The role of ferroptosis in diabetic cardiovascular diseases and the intervention of active ingredients of traditional Chinese medicine.Front Pharmacol. 2023 Oct 26;14:1286718. doi: 10.3389/fphar.2023.1286718. eCollection 2023. Front Pharmacol. 2023. PMID: 37954843 Free PMC article. Review.
-
Non-coding RNAs in necroptosis, pyroptosis, and ferroptosis in cardiovascular diseases.Front Cardiovasc Med. 2022 Aug 4;9:909716. doi: 10.3389/fcvm.2022.909716. eCollection 2022. Front Cardiovasc Med. 2022. PMID: 35990979 Free PMC article. Review.
-
Iron in Cardiovascular Disease: Challenges and Potentials.Front Cardiovasc Med. 2021 Nov 30;8:707138. doi: 10.3389/fcvm.2021.707138. eCollection 2021. Front Cardiovasc Med. 2021. PMID: 34917655 Free PMC article. Review.
Cited by
-
Noncoding RNAs regulating ferroptosis in cardiovascular diseases: novel roles and therapeutic strategies.Mol Cell Biochem. 2024 Nov;479(11):2827-2841. doi: 10.1007/s11010-023-04895-w. Epub 2023 Dec 8. Mol Cell Biochem. 2024. PMID: 38064139 Free PMC article. Review.
-
Ovaries of estrogen receptor 1-deficient mice show iron overload and signs of aging.Front Endocrinol (Lausanne). 2024 Feb 23;15:1325386. doi: 10.3389/fendo.2024.1325386. eCollection 2024. Front Endocrinol (Lausanne). 2024. PMID: 38464972 Free PMC article.
-
Evaluating Intermittent Dosing of Aspirin for Colorectal Cancer Chemoprevention.Cancer Prev Res (Phila). 2025 Jun 2;18(6):321-334. doi: 10.1158/1940-6207.CAPR-24-0168. Cancer Prev Res (Phila). 2025. PMID: 40034042 Clinical Trial.
-
The mechanism of ferroptosis and its related diseases.Mol Biomed. 2023 Oct 16;4(1):33. doi: 10.1186/s43556-023-00142-2. Mol Biomed. 2023. PMID: 37840106 Free PMC article. Review.
-
MMP9 drives ferroptosis by regulating GPX4 and iron signaling.iScience. 2024 Jul 30;27(9):110622. doi: 10.1016/j.isci.2024.110622. eCollection 2024 Sep 20. iScience. 2024. PMID: 39252956 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources