

Emerging epigenetic therapies of cardiac fibrosis and remodelling in heart failure: from basic mechanisms to early clinical development
- PMID: 36004821
- PMCID: PMC10202443
- DOI: 10.1093/cvr/cvac142
Emerging epigenetic therapies of cardiac fibrosis and remodelling in heart failure: from basic mechanisms to early clinical development
Abstract
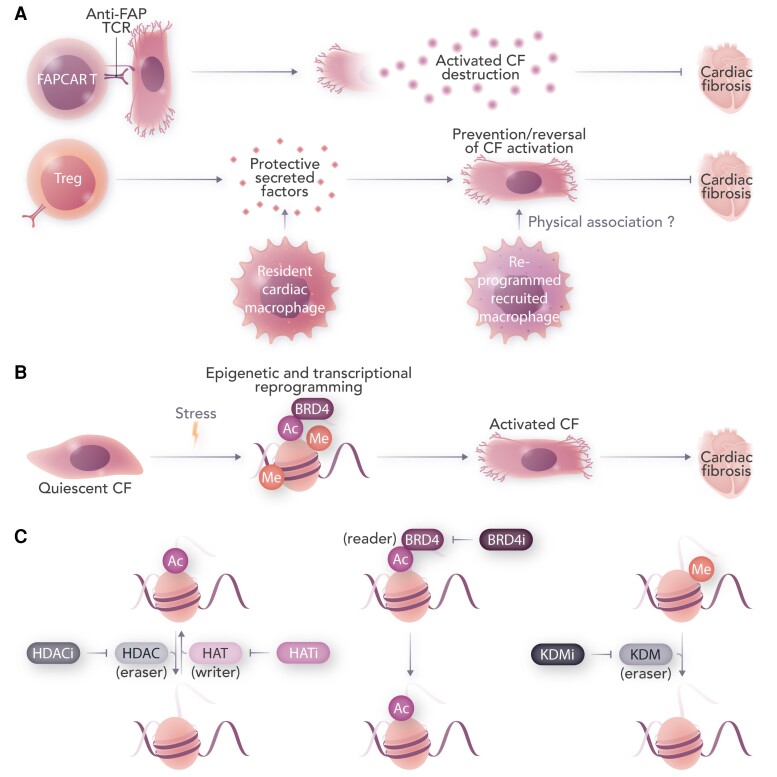
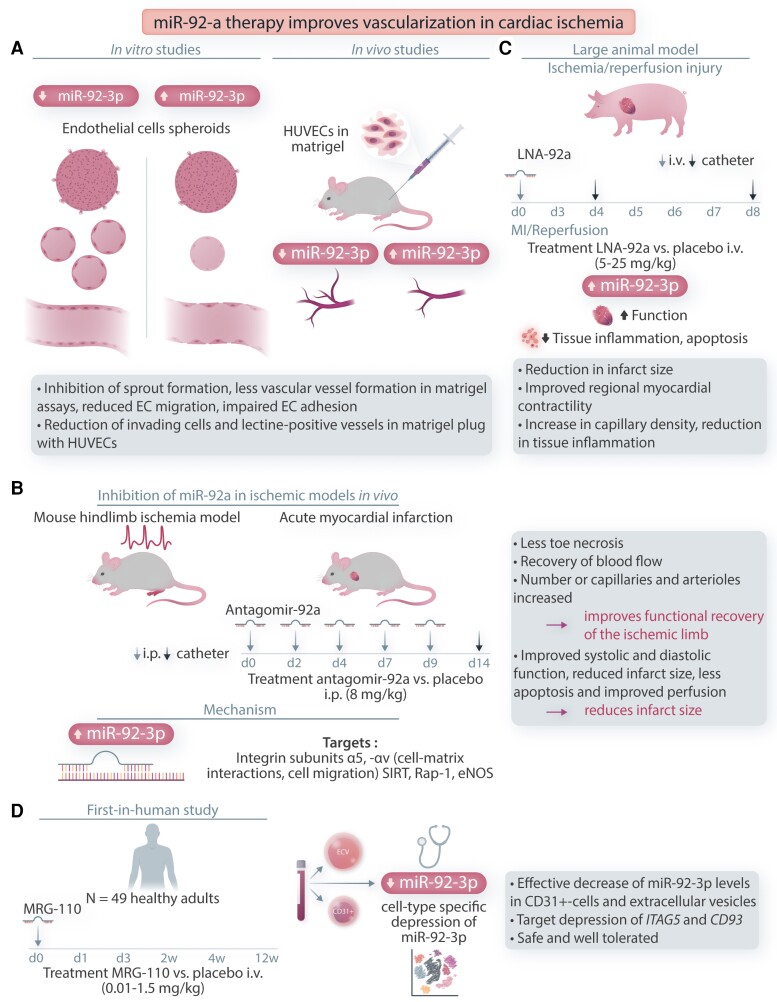
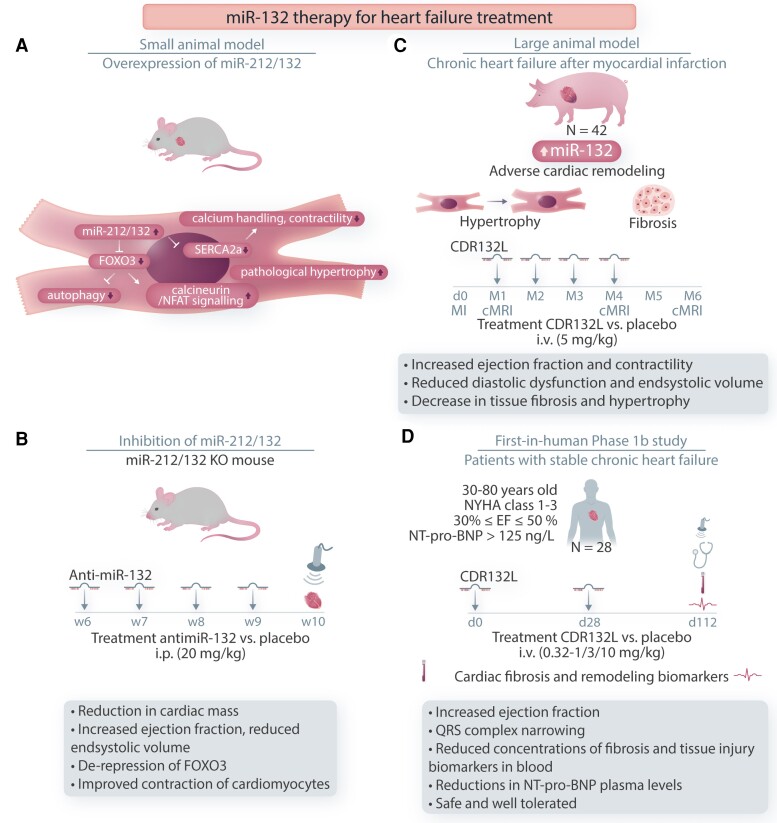
Cardiovascular diseases and specifically heart failure (HF) impact global health and impose a significant economic burden on society. Despite current advances in standard of care, the risks for death and readmission of HF patients remain unacceptably high and new therapeutic strategies to limit HF progression are highly sought. In disease settings, persistent mechanical or neurohormonal stress to the myocardium triggers maladaptive cardiac remodelling, which alters cardiac function and structure at both the molecular and cellular levels. The progression and magnitude of maladaptive cardiac remodelling ultimately leads to the development of HF. Classical therapies for HF are largely protein-based and mostly are targeted to ameliorate the dysregulation of neuroendocrine pathways and halt adverse remodelling. More recently, investigation of novel molecular targets and the application of cellular therapies, epigenetic modifications, and regulatory RNAs has uncovered promising new avenues to address HF. In this review, we summarize the current knowledge on novel cellular and epigenetic therapies and focus on two non-coding RNA-based strategies that reached the phase of early clinical development to counteract cardiac remodelling and HF. The current status of the development of translating those novel therapies to clinical practice, limitations, and future perspectives are additionally discussed.
Keywords: Epigenetics; Fibrosis; Heart failure; MicroRNAs; Non-coding RNAs.
© The Author(s) 2022. Published by Oxford University Press on behalf of the European Society of Cardiology. All rights reserved. For permissions, please email: journals.permissions@oup.com.
Conflict of interest statement
Conflict of interest: T.A.M. is on the scientific advisory board of Artemes Bio and Eikonizo Therapeutics, received funding from Itafarmaco for an unrelated project, and has a subcontract from Eikonizo Therapeutics related to an SBIR grant from the National Institutes of Health (HL15959). T.T. filed and licensed patents in the field of non-coding RNAs and is founder and shareholder of Cardior Pharmaceuticals GmbH. R.F. filed patents in the field of circular RNA and is founder and shareholder of Ephrya Therapeutics (UK).
Figures
References
-
- van den Borne SW, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol 2010;7:30–37. - PubMed
-
- de Jong S, van Veen TA, van Rijen HV, de Bakker JM. Fibrosis and cardiac arrhythmias. J Cardiovasc Pharmacol 2011;57:630–638. - PubMed
-
- Junttila MJ, Holmström L, Pylkäs K, Mantere T, Kaikkonen K, Porvari K, Kortelainen ML, Pakanen L, Kerkelä R, Myerburg RJ, Huikuri HV. Primary myocardial fibrosis as an alternative phenotype pathway of inherited cardiac structural disorders. Circulation 2018;137:2716–2726. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
