

Metabolic Fingerprinting of Fabry Disease: Diagnostic and Prognostic Aspects
- PMID: 36005574
- PMCID: PMC9415061
- DOI: 10.3390/metabo12080703
Metabolic Fingerprinting of Fabry Disease: Diagnostic and Prognostic Aspects
Abstract
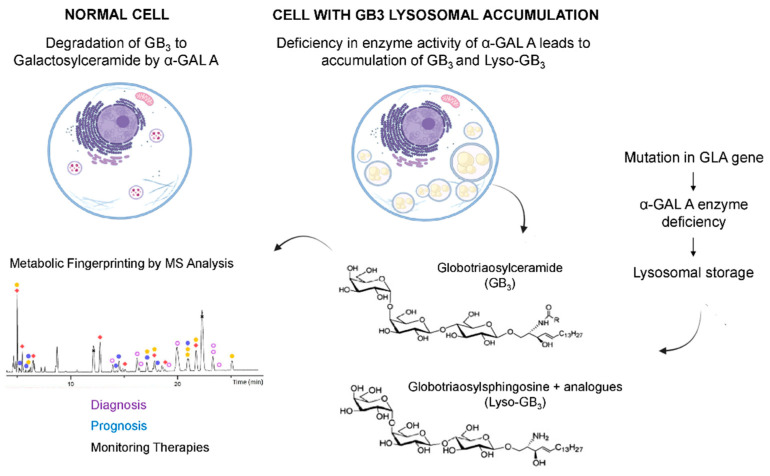
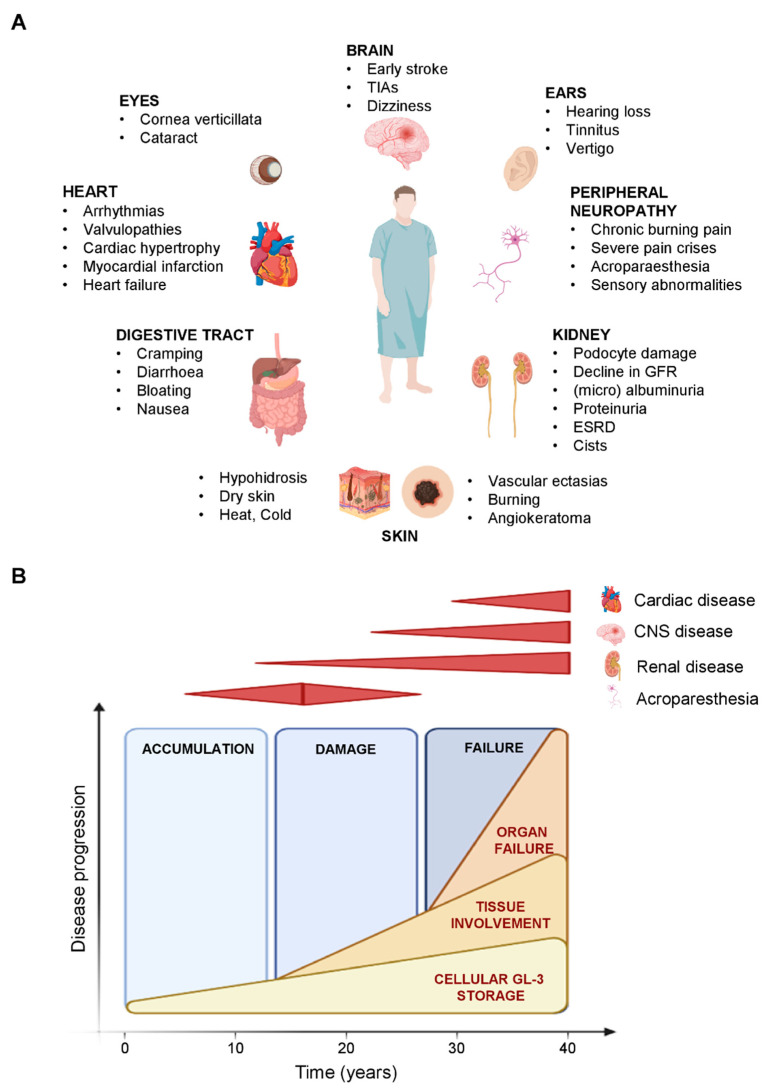
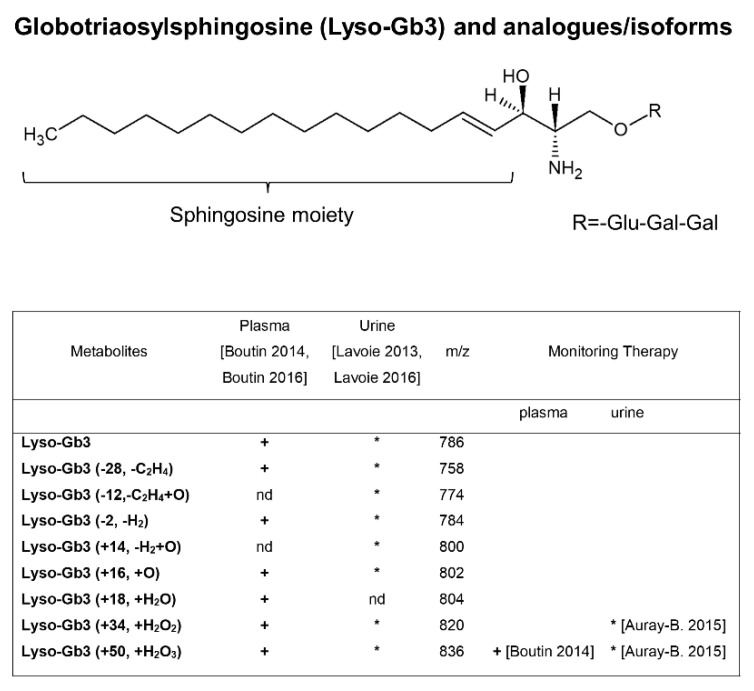
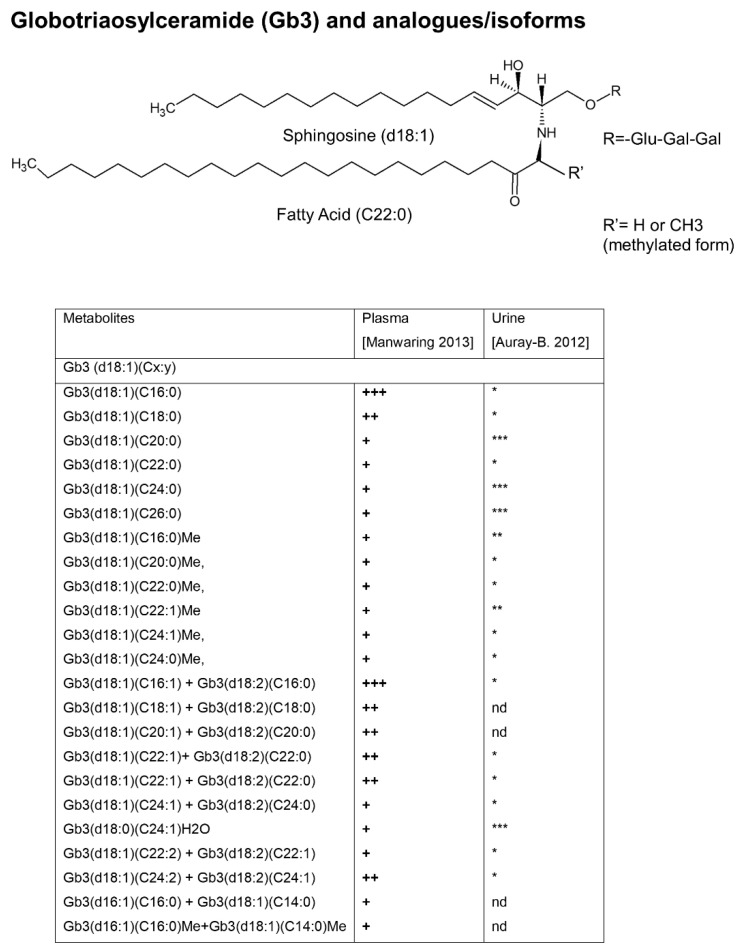
Fabry disease (FD) is an X-linked lysosomal disease due to a deficiency in the activity of the lysosomal-galactosidase A (GalA), a key enzyme in the glycosphingolipid degradation pathway. FD is a complex disease with a poor genotype-phenotype correlation. In the early stages, FD could involve the peripheral nervous system (acroparesthesias and dysautonomia) and the ski (angiokeratoma), but later kidney, heart or central nervous system impairment may significantly decrease life expectancy. The advent of omics technologies offers the possibility of a global, integrated and systemic approach well-suited for the exploration of this complex disease. In this narrative review, we will focus on the main metabolomic studies, which have underscored the importance of detecting biomarkers for a diagnostic and prognostic purpose in FD. These investigations are potentially useful to explain the wide clinical, biochemical and molecular heterogeneity found in FD patients. Moreover, the quantitative mass spectrometry methods developed to evaluate concentrations of these biomarkers in urine and plasma will be described. Finally, the complex metabolic biomarker profile depicted in FD patients will be reported, which varies according to gender, types of mutations, and therapeutic treatment.
Keywords: Fabry disease; LysoGb3; lysosomal storage diseases; metabolomics; systems biology.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Arends M., Wanner C., Hughes D., Mehta A., Oder D., Watkinson O.T., Elliott P.M., Linthorst G.E., Wijburg F.A., Biegstraaten M., et al. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J. Am. Soc. Nephrol. 2017;28:1631–1641. doi: 10.1681/ASN.2016090964. - DOI - PMC - PubMed
-
- Mechtler T.P., Stary S., Metz T.F., De Jesús V.R., Greber-Platzer S., Pollak A., Herkner K.R., Streubel B., Kasper D.C. Neonatal screening for lysosomal storage disorders: Feasibility and incidence from a nationwide study in Austria. Lancet. 2012;379:335–341. doi: 10.1016/S0140-6736(11)61266-X. - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
