

Observation of conformational changes that underlie the catalytic cycle of Xrn2
- PMID: 36008487
- PMCID: PMC9512700
- DOI: 10.1038/s41589-022-01111-6
Observation of conformational changes that underlie the catalytic cycle of Xrn2
Abstract
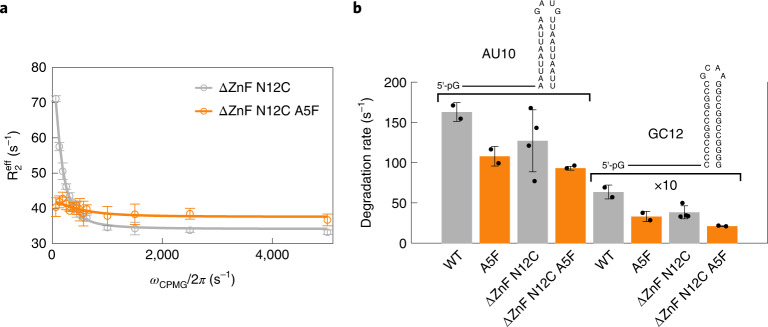
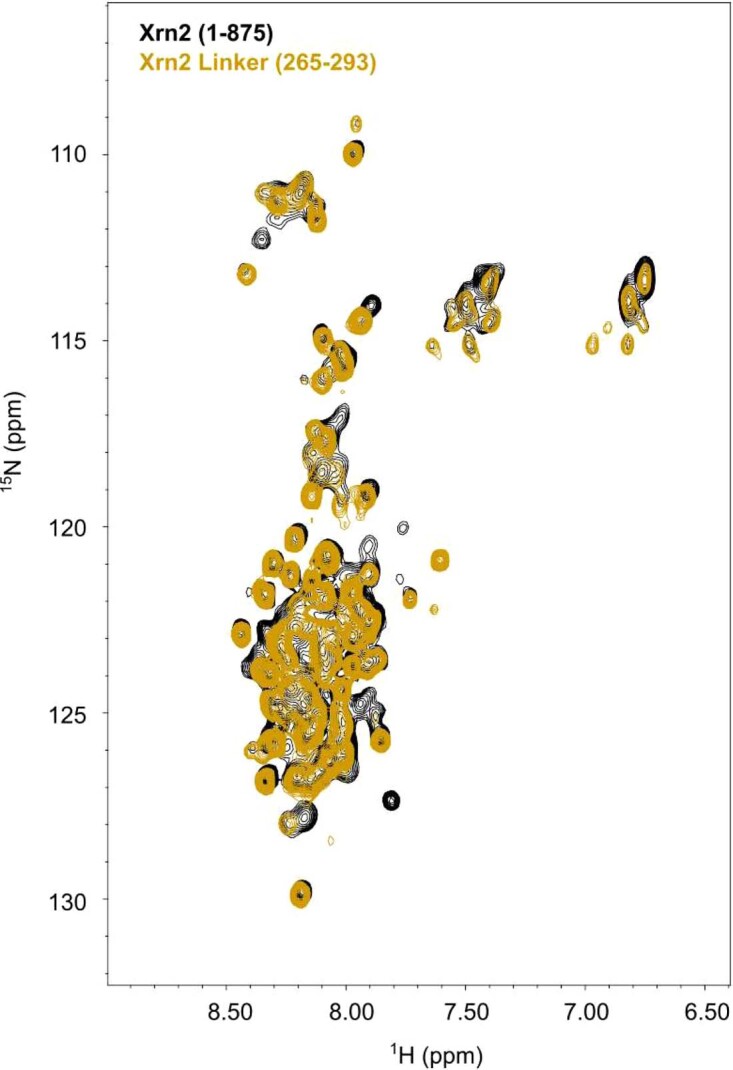
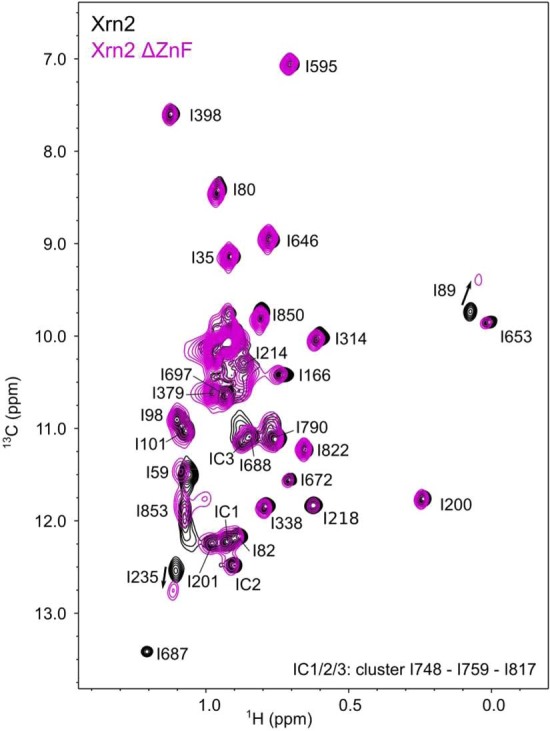
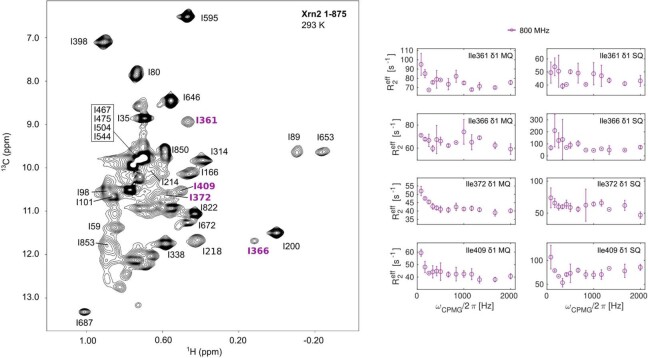
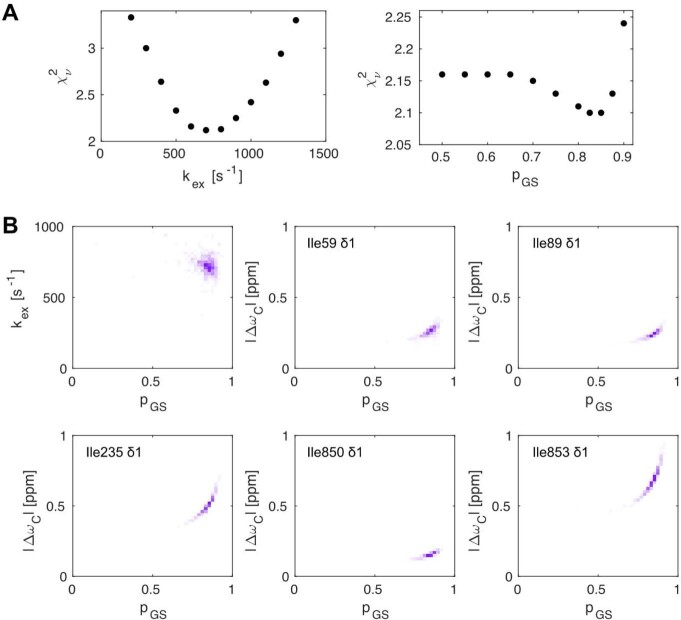
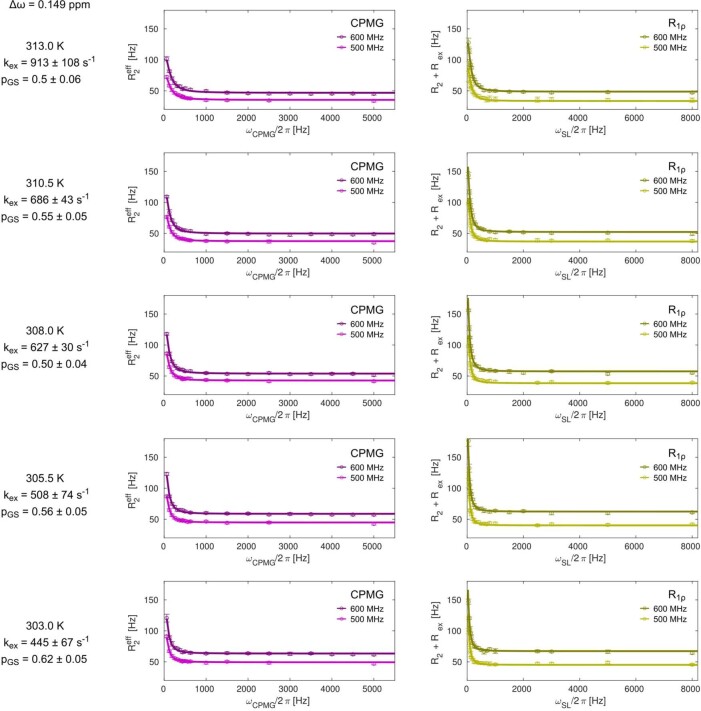
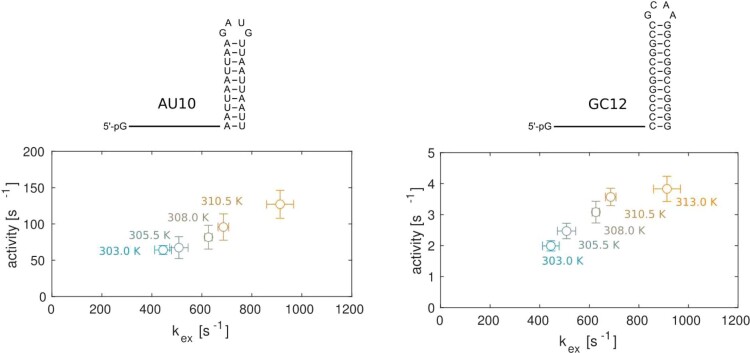
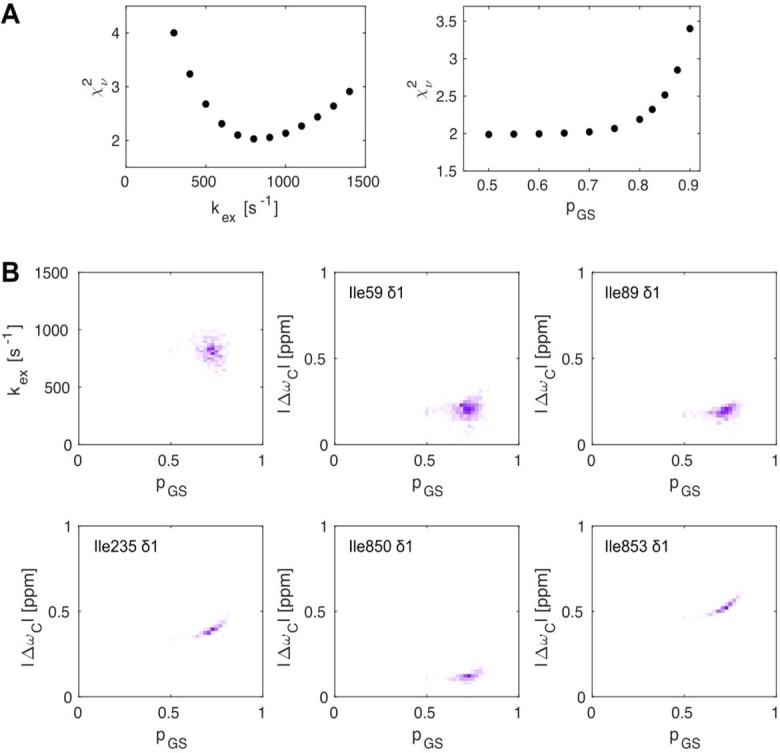
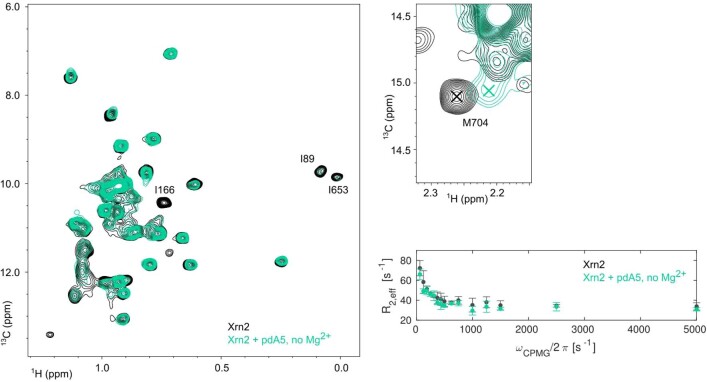
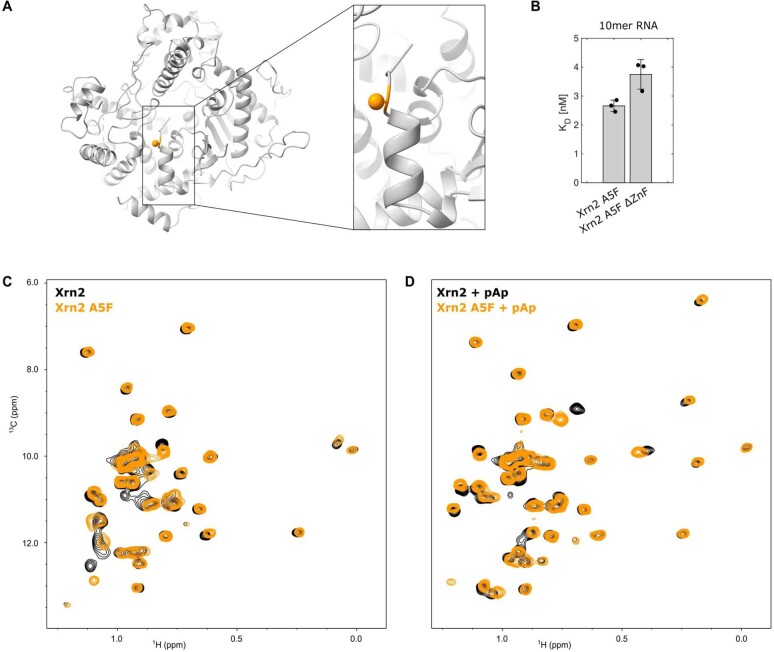
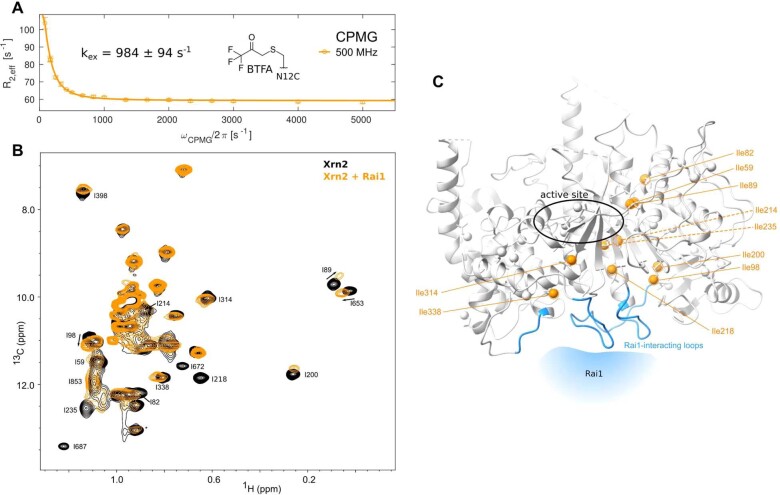
Nuclear magnetic resonance (NMR) methods that quantitatively probe motions on molecular and atomic levels have propelled the understanding of biomolecular processes for which static structures cannot provide a satisfactory description. In this work, we studied the structure and dynamics of the essential 100-kDa eukaryotic 5'→3' exoribonuclease Xrn2. A combination of complementary fluorine and methyl-TROSY NMR spectroscopy reveals that the apo enzyme is highly dynamic around the catalytic center. These observed dynamics are in agreement with a transition of the enzyme from the ground state into a catalytically competent state. We show that the conformational equilibrium in Xrn2 shifts substantially toward the active state in the presence of substrate and magnesium. Finally, our data reveal that the dynamics in Xrn2 correlate with the RNA degradation rate, as a mutation that attenuates motions also affects catalytic activity. In that light, our results stress the importance of studies that go beyond static structural information.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
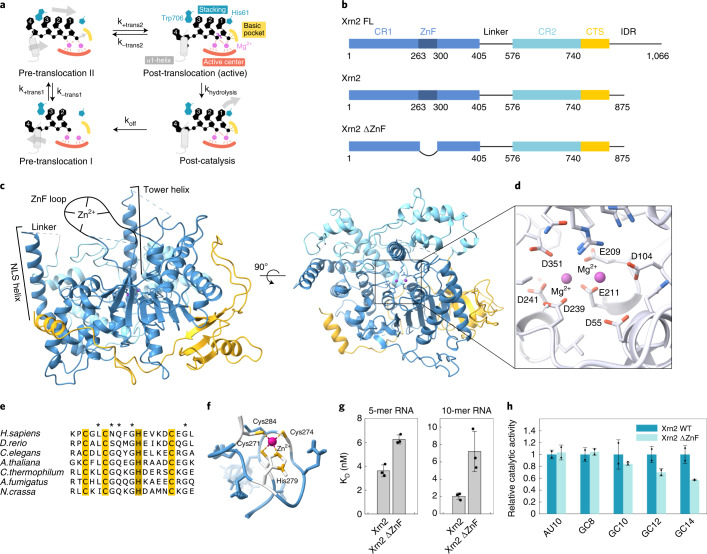
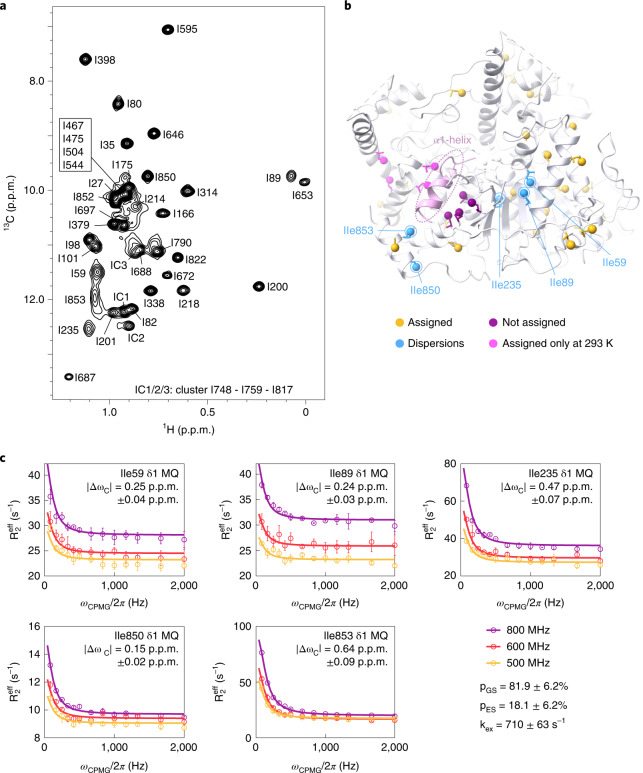
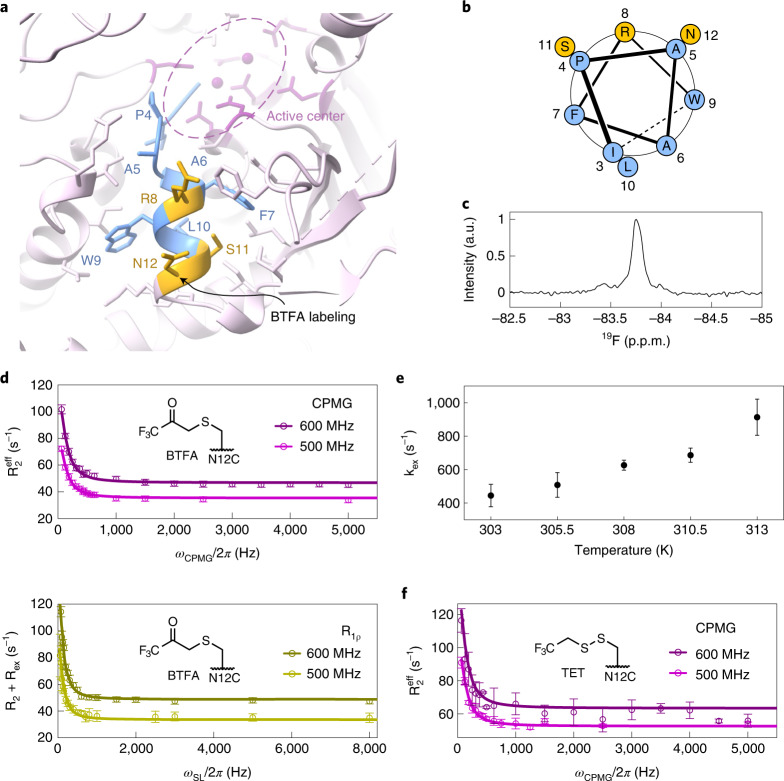
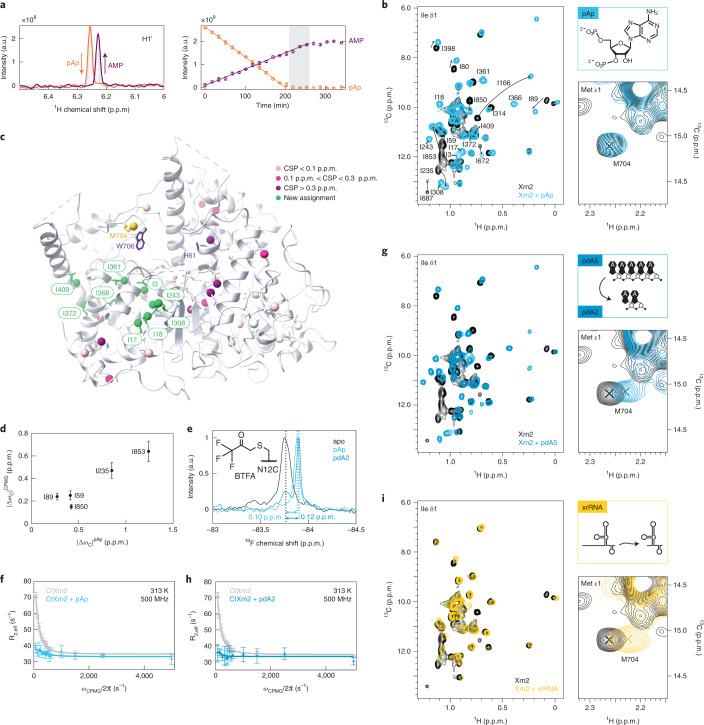
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
